

Latent SLE Presenting as Acute Ascending Paralysis
Latent Systemic Lupus Erythematosus Presenting as Acute Ascending Paralysis: A Case Report
Dr. Kachchakaduge Anushka Peiris 1, Dr. Udari Kaushalya Egodage 2, Dr. Wimalasiri Uluwattage 2
OPEN ACCESS
PUBLISHED: 31 January 2025
CITATION: Peiris, KA., Egodage, UK., et al., 2025. Latent Systemic Lupus Erythematosus Presenting as Acute Ascending Paralysis: A Case Report. Medical Research Archives, [online] 13(1). https://doi.org/10.18103/mra.v13i1.6266
COPYRIGHT: © 2025 European Society of Medicine. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
DOI https://doi.org/10.18103/mra.v13i1.6266
ISSN 2375-1924
Abstract
We report on a 29-year-old mother who presented with acute ascending paralysis who required mechanical ventilation due to severe hypokalaemia. She was found to have normal anion gap metabolic acidosis due to distal renal tubular acidosis. She had ACR and SLICC criteria compatible with diagnosis of latent SLE. SLE is an evolving disease over a lifetime of a patient until it fulfills criteria for clear diagnosis, which makes the management challenging. Early disease may not uncommonly show nonspecific presentation, a single classification criterion, or an unusual organ involvement contributing to frequent, often substantial diagnostic delays. This case is noteworthy for its atypical presentation of SLE. This case demonstrates how unwell and rare a patient with SLE can present. Comprehensive history and examination focusing on systemic and joint symptoms and mucocutaneous involvement, and basic tests (focusing on leukopenia, thrombocytopenia, and proteinuria; followed by antinuclear antibodies and complement levels) will correctly diagnose most patients on presentation or within the following months and enable timely treatment. She was successfully treated with potassium replacement and potassium citrate and sodium bicarbonate supplementation. We also acknowledge the need to further understand and raise awareness of this uncommon presentations in SLE.
Keywords: Hypokalaemia, Distal renal tubular acidosis, Latent Systemic lupus erythematosus, SLICC criteria, Sjogren’s disease
Introduction:
Hypokalaemia is a recognized cause of acute paralysis. Normal anion gap metabolic acidosis secondary to distal renal tubular acidosis could lead to severe hypokalaemia. Defective renal acid-base transporters are described in renal tubular defects. Connective tissue disorders can cause secondary distal renal tubular acidosis. Systemic lupus erythematosus (SLE) is a chronic systemic autoimmune disease with multisystem involvement characterized by the presence of nuclear autoantibodies, a tendency for flare. Rare, atypical, and unusual presentations can involve almost every organ and system, and thus, present to physicians in every discipline and setting. We like to report on a rare presentation of latent SLE with acute ascending paralysis due to severe hypokalaemia secondary to distal renal tubular acidosis. Increasing physicians’ awareness of the potential of occult SLE to appear in varied, diverse, and unexpected presentations, may encourage the inclusion of SLE in the differential. Early recognition and intervention appear to be associated with decreased morbidity.
Case presentation:
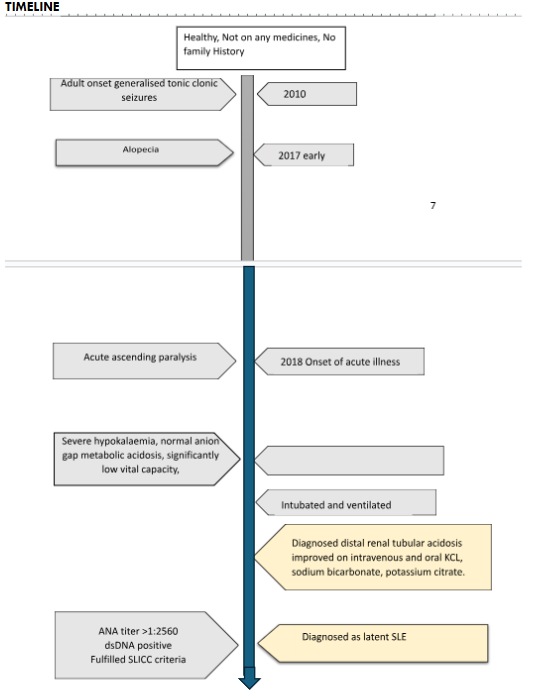
A 29-year-old mother of an infant presented to us with ascending paralysis for three days. She has developed lower limb paralysis initially and progressed to upper limb paralysis. On admission she had difficulty in breathing. She had a history of lower respiratory tract infection a month ago. She has never had previous episodes of paralysis. This episode has not been associated with exercises or meals. She has had recurrent generalized tonic clonic seizures at 21 years which has well responded to antiepileptics. She had noticed excessive hair loss but had no oral ulcers, photosensitivity, facial rashes or joint pains. She had dry mouth but had no dry eyes nor vaginal dryness causing dyspareunia. She had no family history of muscle paralysis.
On admission, she was dyspnoeic and afebrile. Her vital capacity was reduced to 4 mL/kg. All the peripheral reflexes were absent. Bilateral plantar reflexes were flexor. She had proximal muscle weakness of MRC power grade two in both lower limbs and upper limbs. Both fundi were normal. Bilateral cranial nerves were normal. There were no cerebellar signs. She had no sensory impairment. Bladder and bowel were continent. She needed mechanical ventilator support on the first day of admission.
She had severe hypokalaemia (serum potassium 1.1 mmol/L, with normal anion gap metabolic acidosis (ABG pH 7.12, calculated anion gap= 10.0). Corrected serum ionized calcium was 2.31 mmol/L. Serum Magnesium was 1.24mg/dL. Urine pH was 7.2. Urine for sugar was nil. Urine sodium was 71 mmol/L. Urine potassium was 15 mmol/L. Urine calcium to creatinine ratio was 0.09 mg/dL. Urine protein to creatinine ratio was 241.4 g/g revealing sub-nephrotic range proteinuria. Her serum creatinine was 67 umol/L. Xray KUB and the USS KUB revealed no evidence of nephrocalcinosis.
Her hemoglobin was 13.4 g/dL with blood picture revealing normocytic normochromic anaemia. Direct Coomb’s test was positive (DAT positive, Ig G +, Ig G+C3d +, IAT negative). Creatinine Kinase (612 u/l) was Elevated. Her ESR was 129 mm/ 1st hour and C-reactive protein level was 8 mg/L. ANA titer was > 1:2560 with anti-dsDNA negativity. Serum C3 was 134 mg/dL but C4 was l11mg/dL. Rheumatoid factor was 367.3 IU/mL. Anti-Ro and anti-La antibodies were positive. Anti-U1RNP was 4.9 U/mL. Tear break test was normal (right eye 70 seconds, left eye 90 seconds). TSH 3rd Generation was 1.927uiu/ml. CSF analysis done at day 10 of illness onset did not reveal cytoprotein dissociation. Nerve conduction studies were normal except for mild F waves abnormalities.
Her acute condition was treated with intravenous and oral potassium and acidosis correction with sodium bicarbonate and potassium citrate. On second day she has regained her reflexes and could wean off from ventilator, but muscle power was poor. She was successfully mobilized and discharged on oral sodium bicarbonate and potassium citrate after 10 days. Since this patient fulfilled the SLICC criteria for systemic lupus erythematosus including adult onset recurrent seizures and sub-nephrotic range proteinuria under clinical criteria, and positive ANA titer (>1:2560), positive anti-ds-DNA and direct Coomb’s test positivity under immunological criteria we concluded this case as a rare manifestation of latent SLE initially presenting as acute ascending paralysis due to hypokalaemia secondary to distal renal tubular acidosis.
Discussion
Systemic lupus erythematosus (SLE) is a chronic autoimmune disorder of unknown aetiology which may affect any organ of the body. It is a clinically heterogeneous autoimmune disease of unknown etiology. It is characterized serologically by autoantibodies that target self-proteins. Organs and cells of affected individuals undergo damage mediated by tissue-binding autoantibodies and immune complexes. Although the precise etiology of SLE remains vague, genetic predisposition and environmental and hormonal factors are deemed to play important roles in its pathogenesis. SLE patients presents with variable clinical features. Some SLE patients presents with single organ involvement and they may subsequently develop the characteristic multisystem features of SLE over a period of months to years. SLE first presenting as hypokalaemic paralysis due to distal renal tubular acidosis (dRTA) is very rare.
dRTA is characterized by hypokalaemia, normal anion gap metabolic acidosis with inability to lower the urinary pH below 5.5 and could be complicated with acidemia nephrocalcinosis and nephrolithesis. Our patient developed paralysis due to severe hypokalaemia secondary to dRTA. Spot urinary potassium loss of > 20 mmol/day with hypokalaemia is suggestive of renal loss of potassium. Comparing to hypokalaemia secondary to potassium shift into the cells, excessive renal or gastrointestinal loss of potassium needs higher doses of potassium replacement.
Secondary dRTA occurs in autoimmune connective tissue diseases like SLE, Sjogren’s and rheumatoid arthritis. In literature mutations in genes encoding renal acid-base transporters have been suggested in the pathophysiology including mainly luminal membrane H+- K+ ATPase transporters. dRTA in connective tissue diseases is described in literature as due to interstitial nephritis due to mononuclear cell infiltration, deposition of antigen-antibody complexes along tubular basement membrane and antibody formation against tubular basement membrane.
Since our patient fulfilled the 2012 SLICC criteria for SLE but not the American College of Rheumatology criteria for diagnosis of SLE we labelled our patient as a latent SLE who may manifest other multisystemic clinical features overtime. She had to satisfy at least four of the 17 criteria, including at least one of the 11 clinical criteria and one of the six immunologic criteria, or have biopsy-proven nephritis compatible with SLE in the presence of ANA or anti-dsDNA antibodies. Our patient fulfilled five of 17 ANA-positive criteria. In a Scandinavian study of confirmed SLE cases and individuals with lupus-mimicking conditions, the SLICC classification showed comparable diagnostic sensitivity, specificity, and accuracy to the latest classification of European League Against Rheumatism/American College of Rheumatology. ANAs have a sensitivity of 33.6%, while anti-dsDNA has a sensitivity of 57.1%, indicating that a significant portion of SLE cases may be missed based on these markers alone.
Literature says there can be an overlap with SLE and Sjogren’s with anti-Ro and anti-La antibodies positivity. Our patient had dry eyes with anti-Ro and anti-La antibodies positivity but had no other clinical features in mouth with negative tear break test nor typical histological findings on minor salivary gland biopsy to fulfill the diagnostic criteria for Sjogren’s syndrome.
Long-term correction of hypokalaemia is important for reversal of neuromuscular impairment and prevention of renal function deterioration. It has been described that hypokalaemia predisposes deposition of C3, C5ba in the interstitium which aggravates renal interstitial damage. Supplementation of potassium citrate is important for nephrocalcinosis prevention.
We like to highlight the importance of investigating patients with dRTA for treatable secondary cause to minimize complications of an unravelled disease. This case report aims to increase awareness among primary care doctors, who should always consider SLE as a diagnosis in patients presenting with systemic symptoms regardless of absence of full qualification for SLE diagnosis. The shared care and collaboration between different disciplines and levels of care are also important to overcome uncertainties and difficulties in the clinical diagnosis and management of SLE. This is crucial because it leads to early recognition, diagnosis, and treatment, which, in turn, reduces morbidity and increases the quality of life of patients.
Conclusion:
Atypical presentations of SLE could be life threatening. SLE is an evolving disease over a lifetime which makes the diagnosis and management challenging. Further understanding and raising awareness of this uncommon presentations in SLE would benefit this medical space.
References:
- Ter Meulen C.G, Pieters GFFM, Huysmans FTM. Flaccid paresis due to distal renal tubular acidosis preceding systemic lupus erythematosus. Neth J Med 2002; 60:29-32
- Bagga A, Jain Y, Srivastava RN, Bhuyan UN. Renal tubular acidosis preceding systemic lupus erythematosus. Pediatr Nephrol 1993; 7: 735-736.
- DeFranco, P E, Haragsim, L, Schmitz, P G Bastani, B. Absence of vacuolar H(+)-ATPase pump in the collecting duct of a patient with hypokalemic distal renal tubular acidosis and Sjögren’s syndrome. Journal of the American Society of Nephrology: JASN, 1995, 6, 2:295-301
- Gregory A. Kozeny, Walter Barr, Vinod K Bansal, Leonard L Vertuno, Raoul Fresco, John Robinson, Jessie E Hano. Occurrence of renal tubular dysfunction in lupus nephritis. Arch Intern Med 1987; 147: 891- 895.
- Caruana RJ, Barish CF, Buckalew VM Jr. Complete distal renal tubular acidosis in systemic lupus: Clinical and laboratory findings. Am J Kid Dis 1985; 6: 59-63.
- Nakhoul F, Plavnic Y, Lichtig H, Better OS. Hypokalemic flaccid paralysis as the presenting symptom of autoimmune interstitial nephropathy. Isr J Med Sci 1993; 29: 300-303.
- Ray S, Pillai MGK, Kurian G, Unni VN. Latent systemic lupus erythematosus presenting as hypokalemic paralysis. Indian J Nephrol 2005; 15: 98-100
- M. Vendeloo*, A.L.H.J. Aarnoudse, E.F.H. van Bommel. life-threatening hypokalaemic paralysis associated with distal renal tubular acidosis. Netherlands the journal of Medicine 2011; vol. 69, no 1:35-38
- Dahlstrom O, Sjowall C. The diagnostic accuracies of the 2012 SLICC criteria and the proposed EULAR/ACR criteria for systemic lupus erythematosus classification are comparable. Lupus. 2019; 28:778–82. doi: 10.1177/0961203319846388.
- Dey D, Ofori E, Hutton-Mensah KA, Akutek M, Okine R, Amoaba I, et al. Clinical characteristics of males with systemic lupus erythematosus (SLE) in an inception cohort of patients in Ghana. Ghana Med J. 2019; 53:2–7. doi: 10.4314/gmj.v53i1.1.
- Murphy G, Isenberg D. Effect of gender on clinical presentation in systemic lupus erythematosus. Rheumatology (Oxford) 2013; 52:2108–15. doi:10.1093/rheumatology/ket160.