

Congenital Pulmonary Malformations: Insights & Management
Congenital Pulmonary Malformations: Classification and Pathogenesis
Dr. Mónica Saavedra B. 1,2 and Dr. Ximena Ortega F. 3,4
1.Pediatrician specializing in pediatric respiratory diseases, Roberto del Río Children’s Hospital, Las Condes Clinic. Santiago, Chile.; Assistant Professor, Faculty of Medicine, University of Chile, Santiago, Chile.
2.Pediatric Radiologist, Las Condes Clinic, MEDS Clinic, Santiago, Chile.; Adjunct Associate Professor, Faculty of Medicine, Finis Terrae University, Santiago, Chile.
CITATION: Saavedra MB.,and Ortega,XF., 2025.Congenital Pulmonary Malformations Classification and Pathogenesis Medical. research archieves, [online] 13(7)https://doi.org/10.18103/mra.v13i7.6775
ABSTRACT
Background: Congenital pulmonary malformations (CPMs) are rare developmental anomalies affecting the airways, lung parenchyma, and intrathoracic vasculature. Their reported incidence has increased with the widespread use of prenatal imaging. Although often asymptomatic, some CPMs carry a risk of respiratory complications and malignant transformation.
Objective: To summarize current knowledge of the classification, pathogenesis, diagnosis imaging, and clinical implications of CPMs.
Summary: CPMs result from disruptions in embryonic lung development and involve key signaling pathways such as SHH, WNT, BMP, FGF, and TGF-B. Mutations and transcription factors like NKX2.1 and SOX2, along with genetic alterations (e.g. KRAS, DICER) have been identified in several CPMs. Recent advances fetal images studies and postnatal CT angiography have improved early diagnosis and lesion characterization. Long-term studies reveal that even asymptomatic lesions may progress, with a subset showing malignant transformation.
Conclusion: A multidisciplinary approach is essential for managing CPMs. Understanding their molecular basis and identifying prognostic markers are critical for risk stratification and guiding treatment. Standardized classification and longitudinal studies are needed to optimize outcomes.
Keywords: Congenital pulmonary malformations, CPM, lung development, prenatal imaging, malignancy risk.
INTRODUCTION
Congenital pulmonary malformations (CPMs) represent a diverse spectrum of developmental anomalies that originate from disturbances in the embryogenesis of the airway, pulmonary parenchyma, and thoracic vasculature. Their true incidence remains uncertain due to the absence of standardized registries and uniform diagnostic criteria. Historical data have estimated the prevalence of CPMs at 0.5 to 1.5 cases per 10,000 live births. However, in recent decades, the reported frequency has significantly increased, reaching approximately 4 cases per 10,000 live births, according to EUROCAT data. This rise is more likely attributable to enhanced prenatal detection, facilitated by routine obstetric ultrasound and fetal magnetic resonance imaging (MRI), than to a genuine increase in disease prevalence.
The expanding use of advanced imaging techniques has not only improved the early diagnosis of CPMs but also fueled a deeper understanding of their heterogeneous nature and potential clinical implications. There is controversy about the evolution of CPMs, although asymptomatic malformations have been considered to have a benign evolution, they are now recognized as lesions with variable prognosis, including the potential for postnatal respiratory complications and, in some cases, malignant transformation. These observations have stimulated renewed interest in elucidating the underlying pathogenic mechanisms and refining the classification systems used for these anomalies. In this context, recent insights into the molecular, genetic, and epigenetic alterations associated with CPMs have advanced our understanding of their developmental origins. Numerous signaling pathways, such as SHH, WNT, BMP, FGF, and TGF-β, and transcription factors like NKX2.1, SOX2, HOXB5, and GATA6 play critical roles in lung organogenesis. Disruptions in their temporal or spatial expression can result in different malformations, ranging from cystic lesions to complex hybrid anomalies.
Furthermore, current evidence shows that immunological alterations and environmental factors are involved in the pathogenesis of CPMs. This article aims to provide a comprehensive overview of congenital pulmonary malformations by addressing their classification, embryologic basis, and molecular pathogenesis, while also discussing current diagnostic approaches, radiological features, and long-term clinical implications. With this we seek to highlight the importance of a multidisciplinary approach that integrates developmental biology, genomics, imaging and clinical practice in the management and study of these pathologies.
Molecular Mechanisms Involved in the Pathogenesis of Congenital Pulmonary Malformations
Multiple hypotheses have been proposed regarding the pathogenesis of congenital pulmonary malformations (CPMs). Suggested mechanisms include prenatal infections, disruptions in vascular development, airway obstructions, and genetic alterations affecting key processes in pulmonary morphogenesis. These theories are not mutually exclusive, and their interaction may explain the wide heterogeneity of CPMs and the coexistence of multiple anomalies within a single malformation.
To adequately understand the origin of CPMs, it is essential to comprehend the biological processes involved in normal lung development, the participant molecular factors, and the interactions required for proper organogenesis.
In recent years, several studies have identified several critical signaling pathways involved in lung development, identifying numerous factors with relevant regulatory roles. Among them, the transcription factors FOXA1 and FOXA2 (members of the Forkhead box superfamily), as well as GATA4 and GATA6 (zinc finger transcription factors), stand out for their early expression in the endoderm and induction of genes such as SHH. The SHH pathway is critical for anterior foregut differentiation and pulmonary bud formation and regulates the expression of NKX2.1 (also known as TTF-1, thyroid transcription factor-1), which is essential for the development of the pulmonary primordium. The SHH pathway also modulates the expression of bone morphogenetic proteins (BMPs) and WNT2/2B genes, which are key components for bronchial branching.
Notably, BMP4 inhibits the transcription factor SOX2, which is involved in esophageal development, thereby promoting NKX2.1 expression and pulmonary lineage specification.
Additionally, other factors actively participate in lung morphogenesis and airway branching, including fibroblast growth factors (FGFs), the transforming growth factor-beta (TGF-β), and the transcription factor HOXB5, which plays a crucial role during the pseudoglandular stage of lung development and ceases expression after bronchial branching is complete.
Furthermore, effectors of the Hippo signaling pathway—particularly YAP (Yes-associated protein) and TAZ (Transcriptional coactivator with PDZ-binding motif) have shown to modulate lung growth, bronchial branching, and cellular differentiation.
Matrix metalloproteinases (MMPs), especially MMP-7 and MMP-9, have also been found overexpressed in certain CPMs. These enzymes, responsible for degrading components of the extracellular matrix, may alter the cellular microenvironment and contribute to the pathogenesis of these anomalies.
It is worth emphasizing that the expression of molecular factors must be tightly regulated both temporally and spatially during lung morphogenesis. Disruption of these mechanisms can disturb the balance between cellular proliferation and apoptosis, leading to various types of congenital malformations. Research over the past decades has established associations between dysregulation of these pathways and the emergence of several CPM subtypes, some of which are summarized in Table 1.
| Pathway / Factor | Observation |
|---|---|
| BMP | Alteration of BMP leads to cystic lung lesions |
| TBX4-FGF10 | Associated with CPAM Type 0 (acinar dysplasia); overexpression of FGF-10 induces macro and microcystic malformations |
| HOXB5, TTF1, FGF9, and FGF7 | Increased expression of HOXB5, TTF1, and FGF9 and decreased FGF7 have been found in fetal CPAM lesions. Ectopic expression of FGF7 and FGF10 and orthotopic expression of FGF9 disrupt pulmonary morphogenesis |
| HOXB5 | Aberrant expression has been linked to the development of extralobar pulmonary sequestration |
| TTF1 | Altered expression has been observed in CPAM types 1 and 2 and variably in type 4 |
| TGF-β | Dysregulation of TFG-β leads to pulmonary hypoplasia and cystic malformations |
| NKX2.1 and SHH | Mutations cause tracheoesophageal fistulas and pulmonary hypoplasia. |
| SOX2 and TTF1 | SOX2 is absent in adult pulmonary epithelial cells but present alongside TTF1 in cyst cells of CPAM types 1 and 2 |
Genetic and Epigenetic Advances in the Understanding of Congenital Pulmonary Malformations
Recent advances in molecular genetics, particularly the development of technologies such as RNA sequencing, next-generation sequencing (NGS), and DNA methylation analysis, have enabled the identification of significant genetic differences between normal and malformed lung tissues. These techniques have facilitated the detection of gene alterations involved in the genesis of congenital pulmonary malformations (CPMs) but also in the development of certain neoplasms.
One of the most relevant experimental findings has been the overexpression of the SOX2 gene in the pulmonary epithelium of murine models, which disrupts bronchial branching and induces the formation of cystic structures resembling those seen in human congenital pulmonary airway malformation (CPAM). Likewise, both the loss and gain of function of the SOX9 gene have been associated with pulmonary cyst formation, indicating its critical role in lung morphogenesis.
Somatic KRAS gene mutations in CPAM have been identified in the epithelium of cysts. This oncogene is well known for its involvement in several neoplasms, including pancreatic adenocarcinoma and non-small cell lung cancer. The current hypothesis suggests that mucinous proliferations induced by KRAS mutations could develop adenocarcinoma in situ. A study conducted by Hermelijn reported the presence of KRAS mutations in CPAM specimens, supporting the hypothesis that these lesions may have a neoplastic nature.
Another gene of interest is DICER, whose dysfunction has been associated with both abnormal lung development and tumor predisposition. In animal models, FGF10 overexpression induced by DICER alterations results in halted bronchial branching and the formation of large epithelial cystic structures. Additionally, germline variants of the DICER gene have been identified in patients with type I pleuropulmonary blastoma (PPB), suggesting a potential role in pulmonary oncogenesis.
Other genes, such as Activin receptor type II, Lefty1, Nodal, and Pitx2, involved in the regulation of pulmonary symmetry, have also been proposed as potentially implicated in the pathogenesis of certain CPMs. Similarly, overexpression of genes related to the structure and function of ciliated epithelium—DNAH2, DNAH11, CFAP43, CFAP46, RP1, CFAP77, RSPH4A, and DRC3—has been reported in CPMs. This could suggest an alteration in epithelial differentiation mechanisms.
Current research is also focused on identifying potential genetic markers of CPMs and risk markers for malignant transformation. In this context, the SMAD6 gene, a negative regulator of the TGF-β signaling pathway, as well as KRAS-dependent genes, have been proposed as candidates with diagnostic or prognostic value. In this direction, van Horik et al. studies have explored the predictive value of various molecular markers (including MUC1, SOX2, FTT1, and RALDH-1), which could help identify CPM patients at higher risk of developing pulmonary neoplasms.
In the field of epigenetics, DNA methylation alterations, particularly in regions near genes associated with embryonic development and cell proliferation, may also play a role in the genesis of CPMs. This was demonstrated in a study by Patrizi et al., which identified methylation abnormalities previously described in lung tumors and in PPB within a series of congenital pulmonary malformations.
A study conducted by Rodrigues de Moura et al. evaluated genomic instability in resected malformed lung tissue from children with CPMs. Moreover, genetic variants associated with tumors were identified in approximately 30% of the cases, being mucin genes the most frequently mutated group. These findings support the hypothesis of a potential link between CPMs and malignant transformation processes.
OTHER FACTORS INVOLVED IN LUNG MORPHOGENESIS AND THE PATHOGENESIS OF CONGENITAL PULMONARY MALFORMATIONS
RETINOIC ACID (RA):
Retinoic acid (RA), the active form of vitamin A, is essential for lung morphogenesis. It regulates gene transcription via nuclear receptor activation participating in key developmental pathways such as SHH, WNT, and BMP. Several studies have demonstrated that the administration of retinoic acid enhances bronchopulmonary branching in a dose-dependent manner, by modulating genes such as CYP26A1, SOX2/9, RARβ, FGF10, and SHH. Vitamin A deficiency or receptor mutations are linked to severe malformations, including pulmonary hypoplasia and agenesis.
INFLAMMATION AND IMMUNOMODULATION:
Altered innate immune responses, characterized by downregulation of MHC, NK cell, IL-12, and IFN-γ pathways, appear to play a role in the pathophysiology of certain CPMs. These findings suggest a dysfunctional or altered immunological environment in malformed tissue. In this context, treatment with dexamethasone show therapeutic benefit by inhibiting NF-Kβ activity, whereas studies with non-steroidal anti-inflammatory drugs like notrofen may activate pro-inflammatory pathways, contributing to hypoplasia in experimental models.
Imaging Diagnosis and Classification of Congenital Pulmonary Malformations
Currently, there is no universally accepted classification system for congenital pulmonary malformations (CPMs). Definitive identification of these entities requires histopathological examination, which allows for an accurate diagnosis. Nevertheless, advancements in imaging techniques now enable detailed characterization of anomalies, which is essential for appropriate clinical and therapeutic management.
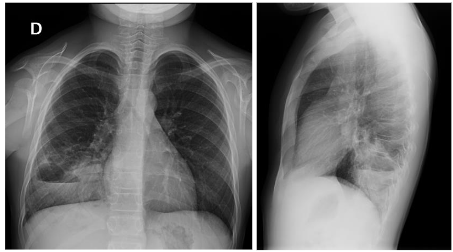
Traditionally, CPMs were diagnosed in the neonatal or postnatal period using chest radiography (Figure 1) or computed tomography (CT) in newborns with unexplained respiratory distress, or in children with recurrent respiratory infections. In other cases, they were incidentally discovered during imaging performed for unrelated reasons.
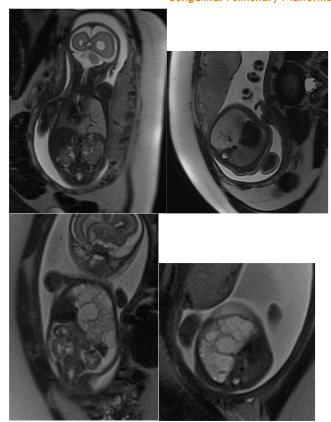
With the advent and widespread use of prenatal ultrasound, it has become possible to detect these malformations as early as 18–20 weeks of gestation. Additionally, fetal magnetic resonance imaging (fetal MRI) has emerged as a valuable complementary tool, providing detailed information about CPMs without the need for contrast agents. It allows for assessment of the size and internal characteristics of the lesion (solid, cystic, or mixed), the presence and course of abnormal vessels from both the pulmonary hilum and systemic circulation, and estimation of the volume of healthy residual lung parenchyma.
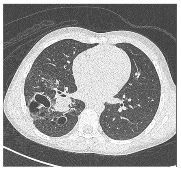
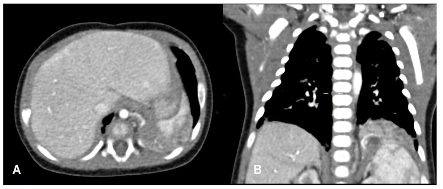
Figure 2. Congenital pulmonary malformations with large and small cystic areas of hyperinflation. No systemic irrigation was observed.
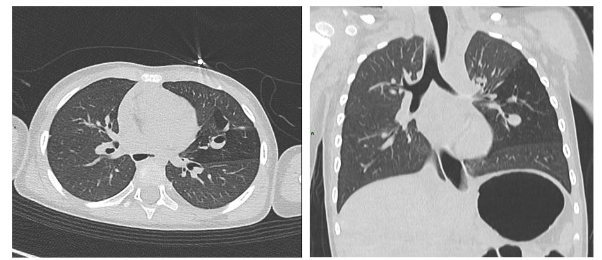
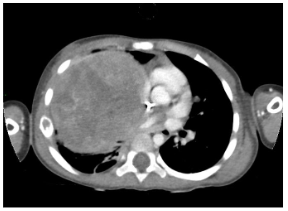
In the postnatal period, the gold standard remains contrast-enhanced computed tomography (CT angiography), which enables high-resolution evaluation of the bronchial tree integrity, characterization of parenchymal architecture (Figure 4), and assessment of the number and size of cysts (Figure 2). It also allows for precise visualization of aberrant systemic vessels (Figure 5), as well as evaluating compressive effects on mediastinal structures.
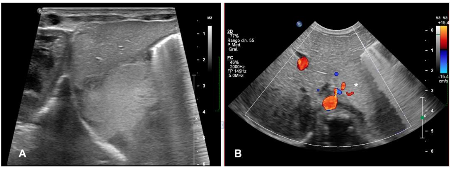
Ultrasound has a limited role in postnatal assessment due to the presence of air in the lungs, which hampers the visualization of deep structures, however, it remains useful in monitoring infradiafragmatic sequestrations.
Postnatal magnetic resonance imaging (MRI) has gained ground as a radiation-free alternative, particularly relevant in infants and for long-term follow-up. High-resolution MRI can allow postponement of CT angiography in asymptomatic neonates until surgery, thereby avoiding early exposure to ionizing radiation. The introduction of ultrashort echo time (UTE) and zero echo time (ZTE) sequences has overcome traditional limitations of thoracic MRI by allowing visualization of aerated lung parenchyma. Furthermore, dynamic MRI sequences have opened a new dimension in functional assessment by enabling ventilation and perfusion studies, which are particularly useful when functional impairment exceeds what is suggested by structural findings.
Regarding the classification of MPC, there is no standard nomenclature, and different classifications have been proposed, among which is the Langston classification, which is based on the pathological characteristics of the lesions, as shown in Table 2.
| Lesion Type | Subtype / Associated Features |
|---|---|
| Bronchogenic cyst | Non-communicating bronchopulmonary foregut malformation |
| Bronchial atresia | -Isolated -With systemic arterial/venous supply (intralobar sequestration) -With gastrointestinal tract communication (intralobar sequestration / complex or communicating bronchopulmonary foregut malformation) |
| Systemic arterial connection to normal lung | |
| Cystic adenomatoid malformation (CAM), large cyst type (Stocker type 1) | -Isolated -With systemic arterial/venous supply (hybrid lesion / intralobar sequestration) |
| Cystic adenomatoid malformation (CAM), small cyst type (Stocker type 2) | -Isolated -With systemic arterial/venous supply (hybrid lesion / intralobar sequestration) |
| Extralobar sequestration | -Without gastrointestinal communication (with/without small cyst-type CAM) -With gastrointestinal communication (complex / communicating bronchopulmonary foregut malformation) |
| Pulmonary hyperplasia and related lesions | -Laryngeal atresia -Solid or adenomatoid CAM (Stocker type 3) -Polyalveolar lobe -Congenital lobar overinflation |
| Other cystic lesions | -Lymphatic/lymphangiomatous cysts -Enteric cysts -Mesothelial cysts -Simple parenchymal cysts -Low-grade cystic pleuropulmonary blastoma |
From a clinical and morphological standpoint, congenital pulmonary malformations (CPMs) can be grouped into three major categories:
- Bronchopulmonary anomalies
- Vascular anomalies
- Combined anomalies
1. Bronchopulmonary Anomalies
Constitute the most frequent group within CPMs and include the following entities:
- Bronchogenic cyst: Originates from abnormal budding of the ventral foregut or aberrations in branching of the tracheobronchial tree. Typical locations include the subcarinal, right paratracheal, paraesophageal, or hilar regions.
- Bronchial atresia: Refers to the absence or interruption of the bronchial lumen at a lobar, segmental, or subsegmental level, with preserved development of the distal lung parenchyma. The proximal bronchus may become dilated and mucus-filled, forming a bronchocele, while the distal parenchyma exhibits hyperinflation due to collateral ventilation. In some cases, aberrant systemic arterial supply may be observed.
- Congenital pulmonary airway malformation (CPAM): This is the most common CPM and includes a spectrum of cystic and non-cystic lesions characterized by abnormal and disorganized proliferation of elements of the tracheobronchial tree. The most widely used classification is that proposed by Stocker, modified in 2002, which groups these malformations into five types based on embryological origin and macro and microscopic features (see Table 2). Since not all lesions exhibit cysts or adenomatoid tissue, the term “congenital cystic adenomatoid malformation (CCAM)” has been largely abandoned.
2. Vascular Anomalies
- Pulmonary arteriovenous malformations (PAVMs): Defined as abnormal direct communications between pulmonary arteries and veins, bypassing the capillary network. A high proportion of cases are associated with hereditary hemorrhagic telangiectasia (HHT) or Rendu-Osler-Weber syndrome, while others occur sporadically.
3. Combined Anomalies
- Pulmonary sequestration: Characterized by non-functioning lung tissue supplied by systemic arterial circulation, usually from the thoracic or abdominal aorta, and drained via pulmonary or systemic veins. The two main subtypes are:
- Intralobar sequestration (ILS): Located within a normal pulmonary lobe sharing its visceral pleura.
- Extralobar sequestration (ELS): Located outside the functional lung, enclosed in its own visceral pleura.
- Scimitar syndrome: Complex congenital anomaly characterized by partial (or rarely, complete) anomalous pulmonary venous return of the right lung to the inferior vena cava, resulting in a left-to-right shunt. It is frequently associated with ipsilateral pulmonary hypoplasia, pulmonary artery anomalies, and aberrant systemic arterial supply, which may originate from supradiaphragmatic or infradiaphragmatic aortic branches.
Long-Term Evolution and Risks Associated with Congenital Pulmonary Malformations (CPMs)
Most CPMs remain asymptomatic. though clinical manifestations may appear at any stage including fetal hydrops, neonatal respiratory distress, recurrent respiratory infections, pneumothorax and development of cancer later in life. Surgical resection is indicated in symptomatic cases, but the optimal management of asymptomatic malformations remains a topic of ongoing debate. Some authors advocate for a conservative approach. Nevertheless, this approach should be taken with caution because emerging evidence has changed the notion that CPMs are entirely benign. In a large series (n=980) by Pederiva et al., 74.4% of adults with CPMs developed symptoms, and 11.7% of resected lesions contained tumors, primarily adenocarcinomas. Notably, malignancies were identified in 25% of CPAMs, 13.7% of ILS, 9.5% of ELS, and 10% of bronchogenic cysts, even in previously asymptomatic individuals.
Other studies have reported similar findings. Aziz et al. reported that 10% of asymptomatic patients with prenatal diagnosis of CPM developed symptoms or complications during follow-up. Similarly, Nasr et al. documented a 4% incidence of tumors in malformations previously considered benign.
These data suggests that CPMs carry a non-negligible risk of complications, including malignant transformation. Consequently, there is a growing need to improve our understanding of the pathobiology of these lesions, clinical signs identification, radiological features that could predict their progression, and biomarkers capable of stratifying the risk of malignancy.
Discussion
Based on the information presented in this article, we can emphasize that Congenital lung malformations constitute a heterogeneous group of developmental lung anomalies with diverse clinical presentations and prognoses. The embryological origin has not yet been fully elucidated, and the molecular mechanisms involved in both the development and progression of these malformations remain unclear. Various signaling pathways (SHH, BMP, WNT, FGF, TGF- β) and transcription factors (NKX2.1, SOX2, HOXB5 among others) participate in their development, and their dysregulation can lead to various malformation.
Current evidence shows that a significant percentage develop respiratory morbidity, and in some cases, they may undergo malignant transformation. Advances in genetics and epigenetics have identified genes and mutations associated with these malformations, some of which are associated with the risk of neoplastic transformation.
Therefore, it is essential to define the molecular and genetic mechanisms involved in the genesis of CPMs and to work on the development of biomarkers and imaging techniques that will allow for the refinement of strategies to adequately define their prognoses.
We must emphasize that the treatment of asymptomatic CPMs remains controversial. Although some groups recommend a watchful waiting approach, recent data should call this approach into question. The malignant transformation rates detected in up to 25% of resected MPC, together with the findings of Pederiva et al and other authors, suggest that strategies are required as clinical course and imaging findings are insufficient to stratify the risk of complications or malignancy and to guide the surgical decision.
The detection of markers such as SOX2, MUC1, TTF1, and RALDH-1, as well as epigenetic alterations, could help better stratify prognosis and guide decision-making. Furthermore, the contribution of immunomodulatory pathways and environmental factors suggests that CPMs have a multifactorial origin. This opens the door for non-surgical, preventative interventions in the future.
However, several limitations remain. Most of the evidence comes from small, observational studies, and multicenter longitudinal studies are required to evaluate long-term outcomes, especially in asymptomatic patients.
Although early diagnosis has significantly increased through obstetric ultrasound and fetal magnetic resonance imaging (MRI), along with advanced postnatal imaging studies such CT angiography, there remains a need to establish a standardized classification of CPMs, allowing for better comparisons between studies.
Furthermore, the integration of approaches that combine genomics, epigenomics, transcriptomics, and proteomics is required to achieve a more complete understanding of the pathogenesis of CPMs.
In conclusion, MPCs reflect the complex interaction between developmental biology and clinical medicine and requires a multidisciplinary approach encompassing pediatrics, radiology, pathology, surgery, molecular biology, and genetics to optimize outcomes. Future research should prioritize the development of risk stratification models based on molecular and imaging markers and standardize CPMs nomenclature and classification. A thorough understanding of their pathogenic mechanisms and their adequate characterization will not only improve individual prognoses but also provide a model for studying normal lung development and its relationship with respiratory tract pathologies.
Bibliography
- Dolk H. EUROCAT: 25 years of European surveillance of congenital anomalies. Arch Dis Child Fetal Neonatal Ed. 2005 Sep;90(5):F355–F358. doi:10.1136/adc.2004.062810
- Lau CT, Kan A, Shek N, Tam P, Wong KKY. Is congenital pulmonary airway malformation really a rare disease? Result of a prospective registry with universal antenatal screening program. Pediatr Surg Int. 2017;33(1):105–108. doi:10.1007/s00383-016-3991-1
- Wan H, Dingle S, Xu Y, Besnard V, Kaestner KH, Ang SL, et al. Compensatory roles of Foxa1 and Foxa2 during lung morphogenesis. J Biol Chem. 2005;280(14):13809–13816.
- White AC, Xu J, Yin Y, Smith C, Schmid G, Ornitz DM. FGF9 and SHH signaling coordinate lung growth and development through regulation of distinct mesenchymal domains. Development. 2006 Apr;133(8):1507–1517. doi:10.1242/dev.02313. PMID: 16540513
- Kuo CT, Morrisey EE, Anandappa R, Sigrist K, Lu MM, Parmacek MS, Soudais C, Leiden JM. GATA4 transcription factor is required for ventral morphogenesis and heart tube formation. Genes Dev. 1997 Apr 15;11(8):1048–1060. doi:10.1101/gad.11.8.1048. PMID: 9136932
- Hrycaj SM, Dye BR, Baker NC, Larsen BM, Burke AC, Spence JR, et al. Hox5 genes regulate the Wnt2/2b-Bmp4-signaling axis during lung development. Cell Rep. 2015;12(6):903-912.
- Domyan ET, Ferretti E, Throckmorton K, Mishina Y, Nicolis SK, Sun X. Signaling through BMP receptors promotes respiratory identity in the foregut via repression of Sox2. Development. 2011;138(5):971-981.
- Gontan C, de Munck A, Vermeij M, Grosveld F, Tibboel D, Rottier R. Sox2 is important for two crucial processes in lung development: Branching morphogenesis and epithelial cell differentiation. Dev Biol. 2008;317(1):296-309.
- Kuwahara A, Lewis AE, Coombes C, Leung FS, Percharde M, Bush JO. Delineating the early transcriptional specification of the mammalian trachea and esophagus. Elife. 2020;9:e55526.
- Volpe MV, Ramadurai SM, Mujahid S, Vong T, Brandao M, Wang KT, et al. Regulatory interactions between androgens, Hoxb5, and TGF-β signaling in murine lung development. Biomed Res Int. 2013;2013:320249.
- Miao Q, Chen H, Luo Y, Chiu J, Chu L, Thornton ME, et al. Abrogation of mesenchyme-specific TGF-β signaling results in lung malformation with prenatal pulmonary cysts in mice. Am J Physiol Lung Cell Mol Physiol. 2021 Jun 1;320(6):L1158–L1168.
- Volpe MAV, Pham L, Lessin M, Ralston SJ, Bhan I, Cutz E, et al. Expression of Hoxb5 during human lung development and in congenital lung malformations. Birth Defects Res A Clin Mol Teratol. 2003 Oct;67(8):550–556.
- Lange AW, Sridharan A, Xu Y, Stripp BR, Perl AK, Whitsett JA. Hippo/Yap signaling controls epithelial progenitor cell proliferation and differentiation in the embryonic and adult lung. J Mol Cell Biol. 2015 Feb;7(1):35–47.
- Parlak A, Ak Aksoy S, Erçelik M, Tekin Ç, Nazlıoğlu HÖ, Tunca B, Gürpınar AN, et al. Matrix metalloproteinase-7 and matrix metalloproteinase-9 expression is upregulated in congenital lung malformations. Turk J Pediatr. 2025 Feb 20;67(1):31–38.
- Luo Y, Cao K, Chiu J, Chen H, Wang HJ, Thornton ME, et al. Defective mesenchymal Bmpr1a-mediated BMP signaling causes congenital pulmonary cysts. eLife. 2023 Feb 28;12:e83327. doi:10.7554/eLife.83327. PMID: 36864912
- Zhang G, Lou L, Shen L, Zeng H, Cai C, Wu R, et al. The underlying molecular mechanism of ciliated epithelium dysfunction and TGF-β signaling in children with congenital pulmonary airway malformations. Sci Rep. 2024 Feb 28;14(1):4430.
- Gonzaga S, Henriques-Coelho T, Davey M, Zoltick PW, Leite-Moreira AF, Correia-Pinto J, et al. Cystic adenomatoid malformations are induced by localized FGF10 overexpression in fetal rat lung. Am J Respir Cell Mol Biol. 2008 Mar;39(3):346–355.
- Clark JC, Tichelaar JW, Wert SE, Itoh N, Perl AKT, Stahlman MT, et al. FGF-10 disrupts lung morphogenesis and causes pulmonary adenomas in vivo. Am J Physiol Lung Cell Mol Physiol. 2001 Apr;280(4):L705–L715.
- Simonet WS, DeRose ML, Bucay N, Nguyen HQ, Wert SE, Zhou L, et al. Pulmonary malformation in transgenic mice expressing human keratinocyte growth factor in the lung. Proc Natl Acad Sci U S A. 1995 Feb 28;92(5):1851–1855.
- Morotti RA, Gutierrez MC, Askin F, Profitt SA, Wert SE, Whitsett JA, et al. Expression of thyroid transcription factor-1 in congenital cystic adenomatoid malformation of the lung. Pediatr Dev Pathol. 2000 Sep-Oct;3(5):455–461. doi:10.1007/s100249900046. PMID: 11143507
- Taylor B, Rice A, Nicholson AG, Hind M, Dean CH. Mechanism of lung development in the aetiology of adult congenital pulmonary airway malformations. Thorax. 2020 Nov;75(11):1001–1003. doi:10.1136/thoraxjnl-2020-214752.
- Rockich BE, Hrycaj SM, Shih HP, Nagy MS, Ferguson MAH, Kopp JL, et al. Sox9 plays multiple roles in the lung epithelium during branching morphogenesis. Proc Natl Acad Sci U S A. 2013 Nov 19;110(47):E4456–E4464.
- Salgia R, Pharaon R, Mambetsariev I, Nam A, Sattler M. The improbable targeted therapy: KRAS as an emerging target in non-small cell lung cancer (NSCLC). Cell Rep Med. 2021 Jan 19;2(1):100186.
- Hermelijn SM, Wolf JL, den Toom TD, Wijnen RMH, Rottier RJ, Schnater JM, et al. Early KRAS oncogenic driver mutations in nonmucinous tissue of congenital pulmonary airway malformations. Hum Pathol. 2020 Sep;103:95–106.
- Harris KS, Zhang Z, McManus MT, Harfe BD, Sun X. Dicer function is essential for lung epithelium morphogenesis. Proc Natl Acad Sci U S A. 2006 Feb 14;103(7):2208–2213.
- Brcic L, Fakler F, Eidenhammer S, Thueringer A, Kashofer K, Kulka J, et al. Pleuropulmonary blastoma type I might arise in congenital pulmonary airway malformation type 4 by acquiring a Dicer1 mutation. Virchows Arch. 2020 Mar;477(3):375–382.
- Zhang S, Ye C, Xiao J, Yang J, Zhu C, Xiao Y, et al. Single-cell transcriptome profiling reveals the mechanism of abnormal proliferation of epithelial cells in congenital cystic adenomatoid malformation. Exp Cell Res. 2020 Sep 1;396(2):112205.
- Jeon HS, Dracheva T, Yang SH, Meerzaman D, Fukuoka J, Shakoori A, et al. SMAD6 contributes to patient survival in non-small cell lung cancer and its knockdown reestablishes TGF-β homeostasis in lung cancer cells. Cancer Res. 2008 Dec 1;68(23):9686–9692.
- van Horik C, Zuidweg MJP, Boerema-De Munck A, Buscop-Van Kempen M, Brosens E, Vahrmeijer AL, et al. Selection of potential targets for stratifying congenital pulmonary airway malformation patients with molecular imaging: is MUC1 the one? Eur Respir Rev. 2023 Apr 26;32(177):230062.
- Patrizi S, Pederiva F, d’Adamo AP. Whole-genome methylation study of congenital lung malformations in children. Front Oncol. 2021 Nov 24;11:751925.
- Rodrigues de Moura R, Patrizi S, Athanasakis E, Schleef J, Pederiva F, d’Adamo AP. Genomic instability in congenital lung malformations in children. Pediatr Surg Int. 2024 Jan;40(1):3–12.
- Rankin SA, Han L, McCracken KW, Kenny AP, Anglin CT, Grigg EA, et al. A retinoic acid–Hedgehog cascade coordinates mesoderm-inducing signals and endoderm competence during lung specification. Cell Rep. 2016 Jan 12;16(1):66–78.
- Fernandes-Silva H, Vaz-Cunha P, Barbosa VB, Silva-Gonçalves C, Correia-Pinto J, Moura RS. Retinoic acid regulates avian lung branching through a molecular network. Cell Mol Life Sci. 2017 Dec;74(24):4599–4619.
- Zhang G, Cai C, Li X, Lou L, Zhou B, Zeng H, et al. Application of second-generation sequencing in congenital pulmonary airway malformations. Sci Rep. 2022 Feb 17;12(1):2986.
- Kotecha S, Barbato A, Bush A, Claus F, Davenport M, Delacourt C, et al.; ERS Lung Congenital Malformation Task Force. Antenatal and postnatal management of congenital cystic adenomatoid malformation. Paediatr Resp Rev. 2012 Jun;13(2):77–86.
- Bush A, Hogg J, Chitty LS. Cystic lung lesions: prenatal diagnosis and management. Prenat Diagn. 2008 May;28(7):604–611.
- Cavoretto P, Molina F, Poggi S, Davenport M, Nicolaides KH. Prenatal diagnosis and outcome of echogenic fetal lung lesions. Ultrasound Obstet Gynecol. 2008 Dec;32(6):769–783.
- Elders BBLJ, Kersten CM, Hermelijn SM, Wielopolski PA, Tiddens HAWM, Schnater JM, et al. Congenital lung abnormalities on magnetic resonance imaging: the CLAM study. Eur Radiol. 2023 Jul;33(7):4767–4779.
- Pacharn P, Kline-Fath B, Calvo-Garcia M, Linam LE, Rubio EI, Salisbury S, et al. Congenital lung lesions: prenatal MRI and postnatal findings. Pediatr Radiol. 2013 Sep;43(9):1136–1143.
- Hermelijn SM, Dragt OV, Bosch JJ, Hijkoop A, Riera L, Ciet P, et al. Congenital lung abnormality quantification by computed tomography: the CLAQ method. Pediatr Pulmonol. 2020 Nov;55(11):3152–3161.
- Langston C. New concepts in the pathology of congenital lung malformations. Semin Pediatr Surg. 2003 Feb;12(1):17–37.
- Stocher JT, Madewell JE, Drake RM. Congenital cystic adenomatoid malformation of the lung: classification and morphologic spectrum. Radiographics. 1994 Nov;14(6):1317–1327.
- Yang W, Gao Y, Li P, Eckman MH. Should asymptomatic patients with congenital lung malformations undergo surgery? A decision analysis. Pediatr Pulmonol. 2023 Feb;58(2):449–456.
- Pederiva F, Dalena P, Pasqua N, Bresesti I, Testa V, Zirpoli S, et al.; European Congenital Lung Malformation Working Group. Risk of malignant transformation and infections in congenital lung malformations in adults: a systematic review. Eur Resp Rev. 2025 Jan 15;34(183):240014.
- Aziz D, Langer JC, Tuuha SE, Ryan G, Ein SH, Kim PCW, et al. Perinatally diagnosed asymptomatic congenital cystic adenomatoid malformation: to resect or not? J Pediatr Surg. 2004 Mar;39(3):329–334.
- Nasr A, Himidan S, Pastor AC, Taylor G, Kim PCW. Is congenital cystic adenomatoid malformation a premalignant lesion for pleuropulmonary blastoma? J Pediatr Surg. 2010 Jun;45(6):1086–1089.