

Molecular Evolution of SARS-CoV-2 Spike Protein Insights
The Molecular Evolution of the SARS-COV-2 Spike Protein: Study of Amino Acid Substitutions and Types
Mike M Moradian, PhD¹², Michael Sweredoski, PhD¹², Tobias Ivan, BS², Ania Baghoomian, BS², Ruan Ramjit, MD¹²
- Kaiser Permanente Bernard J Tyson School of Medicine, Pasadena, CA
- Southern California Permanente Medical Group, Molecular Genetic & Genomic Pathology Regional Laboratory, Los Angeles, CA
OPEN ACCESS
PUBLISHED: 31 March 2025
CITATION: Moradian, MM., Sweredoski, M., et al., 2025. The Molecular Evolution of the SARS-COV-2 Spike Protein: Study of Amino Acid Substitutions and Types. Medical Research Archives, [online] 13(3). https://doi.org/10.18103/mra.v13i3.6382
COPYRIGHT: © 2025 European Society of Medicine. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
DOI https://doi.org/10.18103/mra.v13i3.6382
ISSN 2375-1924
Abstract
The molecular evolution of SARS-COV-2 has been challenging to predict. Emergence of the Omicron Variant of Concern (VOC) and its sublineages indicated that SARS-COV-2 could evolve more rapidly than previously thought. We analyzed the mutation and amino acid substitution patterns in the spike (S) protein of SARS-COV-2 VOCs to assess how they evolved in response to the selective pressure exerted by both natural immunity and vaccination. Our results indicate less evolutionary constraint on the first part of the S protein, allowing more amino acid substitutions, especially in the NTD, RBD, and subdomain 1. Omicron lineages introduced mutations in the FP and HR1 domains for the first time. The NTD, subdomain 1, and FP domains allowed more radical amino acid substitutions, followed by RBD and HR1, possibly due to their function. There were up to nine conservative and one radical substitutions in the amino acids interacting with the Human ACE2 receptor in the RBM of the Omicron sublineages only, a remarkable departure from the previous VOCs. We show that the molecular evolution of SARS-COV-2 S protein was relatively limited up to the Omicron lineage. The selective pressure from previous VOCs and global vaccination potentially accelerated the emergence of the highly transmissible Omicron lineage. This antigenic drift in Omicron is fueled by a high rate of radical amino acid substitutions in the S1 domains, resulting in positive selection with a high potential to change due to adaptive evolution. However, the conservative nature of changes in the RBM may signal a relative stabilization.
Keywords
- SARS-COV-2
- Spike Protein
- Amino Acid Substitutions
- Omicron Variant
- Evolution
Introduction
The molecular evolution of the coronavirus (SARS-COV-2), which has been responsible for a global pandemic with severe and acute respiratory diseases and syndromes, has been quite remarkable. The SARS-COV-2 has evolved rapidly, resulting in at least 4 significant periods of rapid transmission and spread throughout the world. Although mutations have occurred throughout the entire SARS-COV-2 genome, the mutations in the S gene/spike protein have been instrumental in the transmissibility of the virus. Consequently, several studies have focused on the molecular evolution of the spike protein of the SARS-COV-2, indicating that the spike protein is a variable region. They have also reported many non-synonymous mutations in the S gene, which have resulted in both conservative and radical amino acid substitutions, especially in domains such as NTD and RBD. Of course, the acquisition of basic amino acids such as arginine (R) and Histidine (H) in the fusion peptide has also contributed to more efficient fusion of the S protein to the host ACE2 receptors.
The mutations in the S gene and their impact on the spike protein have been different in the major SARS-COV-2 strains. The D614G substitution, which occurred early in the pandemic and gave rise to a more transmissible virus, was one of the first major evolutionary changes that were quickly selected and fixed in the population. All the subsequent major strains of the SARS-COV-2, including the alpha, delta, and omicron have kept the D614G substitution, making it one of the original/founder substitutions. In contrast to D614G, several mutations have disappeared from the major SARS-COV-2 strains, when one strain took over the previous one as the major strain in the world. For example, mutations Del69/70, Del144, N501Y, and others in alpha were not present in delta and the mutations T19R, Del157/158, P681R, and D950N in delta were not present in omicron. These differences indicated that different lineages of the SARS-COV-2 may have evolved independently over time.
While each of the major strains was more transmissible than their predecessor, they had different mutations in their S gene. This could make it challenging to predict an evolutionary pattern. The S gene’s mutation frequencies have changed over time between each strain, studying these mutation frequencies and their types over time could identify the mutations that have been more effective in the virus’s transmissibility. It could also identify the combination of the mutations in a certain time period that could be directly proportional/responsible for the SARS-COV-2 surges.
Another interesting area of the SARS-COV-2 molecular evolution has been the changes in the amino acids that directly bind/interact with the amino acids on the human ACE2 receptor protein. The substitution N501Y, which was a signature substitution of the alpha strain and has been attributed to this strain’s high transmissibility, is a good example of a substitution in a binding/interacting amino acid on the spike protein. Analysis of the type of amino acid substitutions and their frequency in a certain time period or strain could further identify variable or conserved regions in the spike protein amino acids binding/interacting with the host’s ACE2 receptor. Additionally, the rate of evolution of the SARS-COV-2 was thought to decrease gradually over time before the emergence of the Omicron lineage. Wang et al., showed that the coronaviruses have roughly the same mutation rates as the other Flu viruses, Orthomyxoviridae. However, SARS-COV-2 has shown a different and rather unpredictable evolutionary path so far. The emergence of over 30 mutations in the spike protein of Omicron strains, compared to <10 mutations in the previous lineages could be an indication of higher rates of evolution for SARS-COV-2 due to adaptive evolution.
In this study we performed a comprehensive molecular evolution study of the SARS-COV-2 spike protein using the mutation/nucleotide and amino acid substitution patterns. We determined the conserved and variable domains of the spike protein, using radical and conservative amino acid substitution patterns and frequencies.
Materials and Methods
The nucleotide and amino acid substitution patterns and types in the S protein of the SARS-COV-2 were extracted from the platform outbreak.info has been enabled by unprecedented global genomic sequencing efforts. We developed every element of the application to fully leverage this capacity; however, genomic sampling varies globally with the vast majority of sequences coming from high income countries; even within well-sampled regions, there is geographic and temporal variation. Only the amino acid substitutions that were present in more than 80% of the reported SARS-COV-2 sequences in the outbreak.info database were used in this study. The web application was built using Vue.js v.2.7.14 (https://vuejs.org/), a model–view–view model JavaScript framework that enables the two-way binding of user interface elements and the underlying data allowing the user interface to reflect any changes in underlying data and vice versa. The client-side application uses the high-performance API to perform interactive operations on the database. In addition, All SARS-CoV-2 virus sequence data were provided by the GISAID Global Data Science Initiative and are available at https://gisaid.org/.
In Figure 1, to calculate the expected proportion of mutations per domain, we divided domain length by the total spike protein length. The proportion of mutations in each domain was calculated as the total number of mutations in the domain in the VOC divided by the total number of mutations in the VOC.
The radical or conservative designations were determined by using the Grantham tables/score. A score of 90 or above was designated as radical amino acid substitution and a score of <90 as conservative amino acid substitutions.
Results
DEVIATIONS FROM EXPECTED NUMBER OF MUTATIONS
In the closer VOC linages to Wuhan (i.e., Beta and Gamma), the mutations were concentrated in the NTD, RBD, and subdomain 1 (SD1, the area between RBD and the fusion peptide); all three domains had a higher proportion of mutations than expected. The picture was slightly different in Alpha and Delta, where a higher-than-expected proportion of mutations appeared in the HR1 domain. While in Alpha the proportion of mutations in the RBD was in line or slightly lower than the expectations, a sudden increase in SD1 mutations could have had a compensatory effect on the low mutation frequency in the RBD. However, the emergence of N501Y substitution, which directly interacts with an ACE2 receptor amino acid, along with P681H could have increased the efficiency of the Alpha variant and reduced the need for more RBD mutations. The NTD in Delta has more than twice the expected proportion of mutations, which could have been the reason for Delta’s efficient transmissibility, possibly evading the neutralizing antibodies.
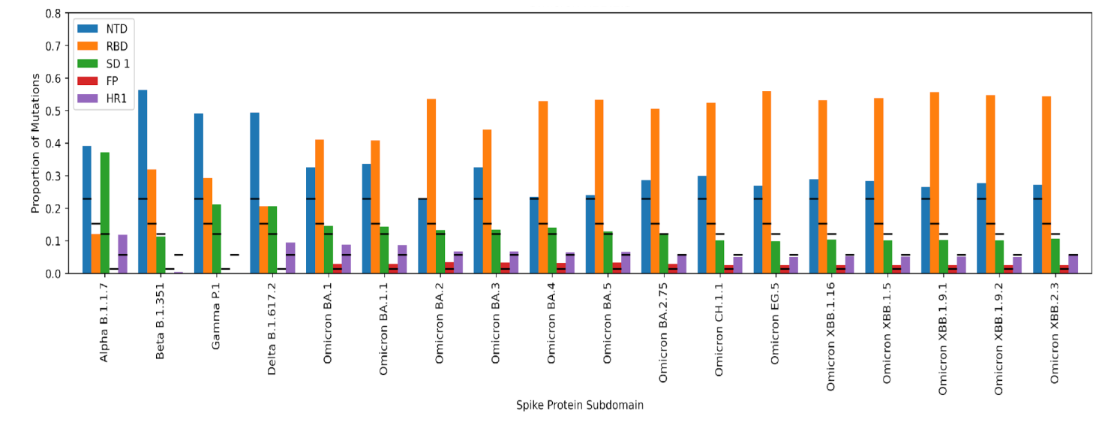
The high proportion of mutations in the Omicron lineage, especially in the RBD was a remarkable shift. For the first time since the pandemic began, in any lineage, there are more than the expected proportion of mutations in the FP domain. Omicron lineages seemingly have a sufficient number of mutations in the NTD to possibly evade the neutralizing antibodies. Furthermore, they have a very high proportion of mutations in the RBD, SD1, FP, and HR1 domains to possibly transmit and infect the host cell more effectively. Also, they have 7-9 mutations in the amino acid positions on the spike protein that interact with the ACE2 receptor amino acids, which could potentially increase the efficiency of their host cell binding and transmission.
MUTATIONS AND AMINO ACID SUBSTITUTIONS IN THE SPIKE PROTEIN
We analyzed the mutations in the spike protein of the VOCs based on their frequency, types, and patterns in each respective VOC. There were significant differences between the number of mutations amongst the VOCs per domain (NTD p-value: 5.7 x 10-2, RBD p-value: 1.8 x 10-9, SBD 1 p-value: 1.8 x 10-2, Fusion Peptide p-value: 1.1 x 10-4, and HR1 p-value: 4.3 x 10-4. While Omicron sublineages had the highest number of mutations, they were not evenly distributed across the S protein.
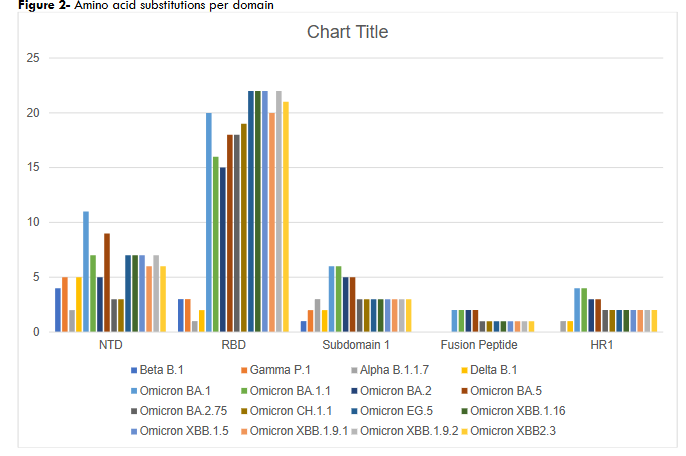
Mutations in Omicron sublineages were concentrated in the RBD (i.e., up to 22 mutations), at least half of which occurred in the RBM. In the NTD, the presence of several amino acid deletions and substitutions indicates several variable positions in this domain. These include amino acid positions 18-27 (Del 25/27 in all Omicrons), positions 67-70 (Del 69/70 in Alpha and BA.2.86), positions 142-158 (Del 144 in Alpha and all Omicrons except CH.1.1, and Del 157/158 in Delta), and the positions 211-215 (Del 212 BA). These deletion events along with several other substitutions indicate that the NTD could tolerate a fair number of changes and may not be under strong evolutionary conservation pressure.
While the other regions/domains in S protein, such as SD1, FP, and HR1 had fewer amino acid substitutions, the numbers were similarly different between VOCs, with more mutations in the Omicron sublineages. The main difference between the Omicron lineages and the rest of the VOCs is in the RBD/RBM. Omicron sublineages have up to 22 mutations in this domain while the rest of the VOCs had 9 combined. Only two of the Omicron mutations in the RBM have been previously reported (K417N in Beta and N501Y in Alpha, Beta, and Gamma), the rest are novel. The N501Y substitution, which is considered a key mutation that increases the transmission of the SARS-COV-2 and is present in almost all the VOCs, was absent in Delta. All Alpha and Omicron lineages (except for BA.2.86) have the P681H substitution in the S1/S2 cleavage site This substitution represents a key difference between the Alpha and Omicron lineages and the Delta and BA.2.86 lineages, which have the substitution P681R.
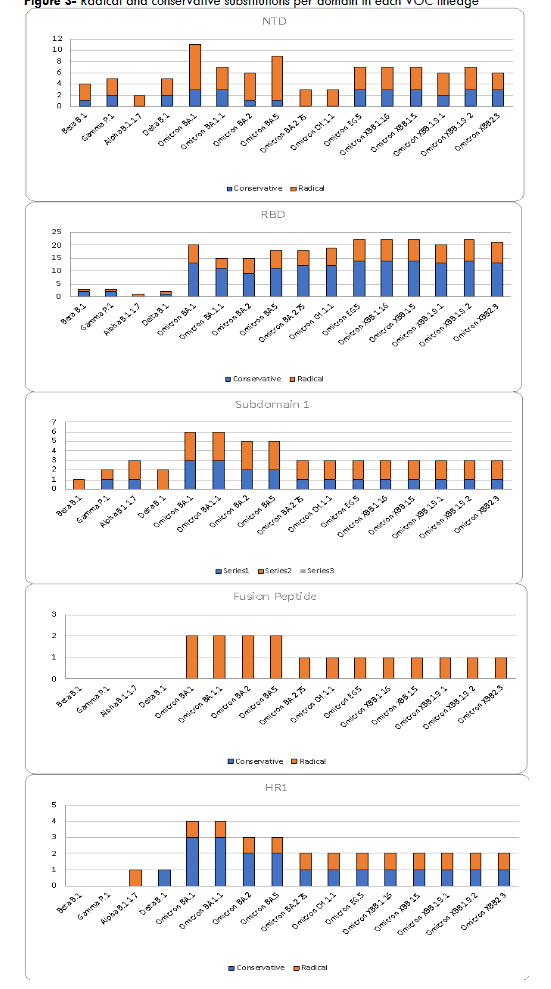
The amino acid substitution patterns showed interesting differences between the VOCs. The VOCs before Alpha, such as Beta and Gamma, which did not spread as rapidly globally, did not have any amino acid deletion in the NTD, while all the VOCs after alpha had at least 2 and up to 5 amino acid deletions, suggesting that these deletions may have played a potentially significant role in their global transmission. We also observed more radical amino acid substitutions across all the VOCs in the NTD than conservative substitutions. In contrast, up to the Omicron lineage the RBD had more conservative substitutions than radical substitutions, which were primarily in the RBM region. Omicron sublineages had radical amino acid substitutions throughout the RBD domain with up to half of them being radical in the RBM, suggesting less conservation across this domain. There were more radical than conservative amino acid substitutions in SD1 and the FP domains. In fact, FP had only radical amino acid substitutions and only in the Omicron sublineages. Except for Alpha, the HR1 domain had more conservative than radical amino acid substitutions in all the VOCs. In Alpha, there was only one amino acid substitution (radical), a distinct difference from the other VOCs.
Discussion
Analysis of the nucleotide and amino acid substitution patterns and types in the S protein of the SARS-COV-2 allowed us to provide further evidence of the molecular evolution of this virus. The amino acid substitution types indicated that the S protein allows two very different patterns of evolution, a highly variable region starting at the NTD to the HR1 domain and a highly conserved region thereafter. Different studies have already suggested the reasons for such structure, yet the emergence of the Omicron lineage has changed the molecular evolution of the SARS-COV-2. The domains in the S1 region of the Omicron lineage proved to be highly variable, possibly resulting in an antigenic drift from previous VOCs. The main difference was in the RBD/RBM between the Omicron sublineages and the rest of the VOCs. This region, which contains the amino acid residues that interact with the ACE2 receptor in human cells, has been quite conserved in the previous VOCs, yet introduction of up to 22 amino acid substitutions in Omicron sublineages completely changed that picture. These nonsynonymous mutations, resulting in amino acid substitutions in the RBD indicate an evolutionary response to the pressure from antibodies due to vaccination and prior infections in SARS-COV-2. Two studies presented 26 amino acids in the RBM that they believed interact with the human ACE2. All the VOCs before Omicron had between zero and three substitutions, Omicron sublineages have up to nine, meaning about one-third of the presumptive interacting amino acids in the Omicron RBM were mutated/substituted. This adaptation could be the reason for Omicron’s supreme transmissibility.
Since the RBD/RBM amino acids interact with ACE2, radical changes—especially in higher numbers—may be disadvantageous to the SARS-COV-2 transmissibility. The adaptive evolution of the S1 domains in the Omicron lineage was not uniform; all domains, except the RBD, had more radical changes/amino acid substitutions. The relative stability (i.e., more conservative amino acid substitutions) in the RBD could be primarily due to the fact that eight out of nine amino acid substitutions in the RBM, which interact with the human ACE2 protein were conservative; the exception was N501Y. The domination of the conservative amino acid substitutions in the RBM is notable, suggesting that there may be a limit to the antigenic drift in this domain. If RBM does not allow radical changes to its structure, then its adaptive evolution may be stabilizing.
Substitutions at residues L452, F486, and R493, which are targets for neutralizing antibodies in the RBM, have been shown to confer resistance in the Omicron sublineages. Although most of the amino acid substitutions in the RBM of the Omicron sublineages were conservative, the BA.5 RBM had three radical substitutions (highest among all Omicron sublineages) from a total of nine. This sublineage has also shown substantially more neutralization resistance to sera obtained from vaccinated and boosted individuals. If there is any correlation between radical amino acid substitutions in the RBM and antibody evasion, then we may anticipate seeing more of them in the future due to vaccine boosters.
The rates of evolution for SARS-COV-2 have been suggested to be in line with other similar respiratory viruses. This was true up to the Delta lineage, yet the Omicron lineage proved that perhaps it is early to be able to estimate the evolutionary rate of the SARS-COV-2 accurately. Another analysis estimated the rate of S1 mutations to be four times that of the influenza H3N2 virus. With such a high drift rate in the antigenic region of the SARS-CoV-2, the need to create vaccine boosters for the new lineages has become a reality. This could create a vicious cycle by putting additional selective pressure on the virus to adapt and evade the new antibodies, consequently, creating new lineages. However, since the pressure would be on the virus’s transmission machinery the pathogenicity of it could remain unchanged. At least this is what was observed with the emergence of the Omicron lineage.
Analysis of the amino acid substitution types and patterns revealed interesting observations. None of the VOCs before Alpha had deletion(s) in their NTD, suggesting that the deletions in the NTD synergize with D614G substitution to enable ACE2-independent S2’ cleavage, which is believed to assist with transmissibility. Also, while these VOCs had the only radical substitution in an interacting amino acid, i.e., N501Y, which increases the ACE2 binding affinity of the S protein, they missed the basic substitutions in position 681, which is a preferred interacting amino acid by furin. The absence of these two important changes in early VOCs may have been a factor in their slower/limited spread in the world. Interestingly, N501Y seems to be present with substitution P681H in more transmittable variants of Alpha and Omicron. While N501Y was not present in the Delta sublineage, the presence of substitution P681R could compensate for this missing mutation, creating the basic cleavage motif in the P5 site. Arginine in position 681 confers the greatest increase in cleavage efficiency. This raises an interesting question and a future opportunity to investigate if there is any connection between the amino acid positions 501 and 681 in the S protein binding and fusion to the human cells.
Overall, adaptive evolution seems to be the driving force for the rapid molecular evolution of the S protein in SARS-COV-2. Accumulation of the nonsynonymous mutations in the S protein of different lineages has been a good indication of that lineage’s success. Also, specific substitutions may have contributed to the lineages’ success more than the others; these substitutions have been fixed in multiple VOCs through convergent evolution. We demonstrated that the remarkable antigenic drift in Omicron sublineages and their effective and persistent presence as the dominant SARS-COV-2 clade for the longest time may be accompanied by the significantly higher rate of radical amino acid substitutions in their S protein. However, these patterns may also point to stabilizing the S protein evolution and less dramatic changes in the spread of the SARS-COV2 throughout the world.
Conclusion
The molecular evolution of the SARS-CoV-2 spike (S) protein has demonstrated a complex interplay between selective pressures, antigenic drift, and adaptive evolution. Our study highlights that while early variants of concern (VOCs) exhibited relatively constrained evolution, the emergence of the Omicron lineage marked a dramatic shift in mutation frequency and distribution. Omicron sublineages introduced novel mutations in key functional domains, including the receptor-binding domain (RBD) and fusion peptide (FP), contributing to increased transmissibility. Our analysis of amino acid substitution patterns indicates that SARS-CoV-2 has undergone significant antigenic drift, particularly in regions associated with antibody recognition and host cell interaction. The high prevalence of radical amino acid substitutions in the S1 subunit suggests that SARS-CoV-2 continues to evolve in response to immune pressure from prior infections and vaccination efforts. However, the relative conservation of the receptor-binding motif (RBM) suggests a potential stabilization of viral-host interactions, which may limit the extent of future antigenic changes in this region.
These findings underscore the dynamic nature of SARS-CoV-2 evolution, with implications for public health strategies and vaccine development. As new variants continue to emerge, close monitoring of spike protein mutations will be critical for anticipating changes in transmissibility, immune escape, and vaccine efficacy. While the rapid accumulation of mutations in Omicron sublineages presents challenges for long-term viral control, the observed conservation in key functional sites may provide opportunities for designing more robust and broadly protective countermeasures against future SARS-CoV-2 variants.
Conflict of Interest Statement
The authors have no conflicts of interest to declare. The authors have no disclosures to report.
References
- Markov PV, Ghafari M, Beer M, et al. The evolution of SARS-CoV-2. Nature Reviews Microbiology. 2023;21(21):1-19. doi:https://doi.org/10.1038/s41579-023-00878-2
- Yajima H, Tomo Nomai, Okumura K, et al. Molecular and structural insights into SARS-CoV-2 evolution: from BA.2 to XBB subvariants. mBio. 2024;15(10). doi:https://doi.org/10.1128/mbio.03220-23
- Huang Y, Yang C, Xu X, Xu W, Liu S. Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacologica Sinica. 2020;41(9):1141-1149. doi:https://doi.org/10.1038/s41401-020-0485-4
- Saito A, Irie T, Suzuki R, et al. Enhanced fusogenicity and pathogenicity of SARS-CoV-2 Delta P681R mutation. Nature. 2021;602:1-10. doi:https://doi.org/10.1038/s41586-021-04266-9
- Wolf KA, Kwan JC, Kamil JP. Structural Dynamics and Molecular Evolution of the SARS-CoV-2 Spike Protein. mBio. 2022;13(2). doi:https://doi.org/10.1128/mbio.02030-21
- Korber B, Fischer WM, Gnanakaran S, et al. Tracking Changes in SARS-CoV-2 Spike: Evidence That D614G Increases Infectivity of the COVID-19 Virus. Cell. 2020;182(4). doi:https://doi.org/10.1016/j.cell.2020.06.043
- Plante JA, Liu Y, Liu J, et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature. 2020;592. doi:https://doi.org/10.1038/s41586-020-2895-3
- Liu Y, Liu J, Plante KS, et al. The N501Y spike substitution enhances SARS-CoV-2 infection and transmission. Nature. Published online November 24, 2021. doi:https://doi.org/10.1038/s41586-021-04245-0
- Volz E, Mishra S, Chand M, et al. Assessing transmissibility of SARS-CoV-2 lineage B.1.1.7 in England. Nature. 2021;593(7858):266-269. doi:https://doi.org/10.1038/s41586-021-03470-x
- Magazine N, Zhang T, Wu Y, McGee MC, Veggiani G, Huang W. Mutations and Evolution of the SARS-CoV-2 Spike Protein. Viruses. 2022;14(3):640. doi:https://doi.org/10.3390/v14030640
- Chen Y, Guo Y, Pan Y, Zhao ZJ. Structure analysis of the receptor binding of 2019-nCoV. Biochemical and Biophysical Research Communications. 2020;525(1). doi:https://doi.org/10.1016/j.bbrc.2020.02.071
- Wang S, Xu X, Wei C, et al. Molecular evolutionary characteristics of SARS‐CoV‐2 emerging in the United States. Journal of Medical Virology. 2021;94(1):310-317. doi:https://doi.org/10.1002/jmv.27331
- Wang Q, Guo Y, Iketani S, et al. Antibody evasion by SARS-CoV-2 Omicron subvariants BA.2.12.1, BA.4 and BA.5. Nature. 2022;608(7923):603-608. doi:https://doi.org/10.1038/s41586-022-05053-w
- Viana R, Moyo S, Amoako DG, et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature. 2022;603(7902):679-686. doi:https://doi.org/10.1038/s41586-022-04411-y
- Kistler KE, Bedford T. Evidence for adaptive evolution in the receptor-binding domain of seasonal coronaviruses OC43 and 229e. eLife. 2021;10(33463525). doi:https://doi.org/10.7554/elife.64509
- Gangavarapu K, Latif AA, Mullen JL, et al. Outbreak.info genomic reports: scalable and dynamic surveillance of SARS-CoV-2 variants and mutations. Nature Methods. 2023;20(4):512-522. doi:https://doi.org/10.1038/s41592-023-01769-3
- Tseung G, Mullen JL, Alkuzweny M, et al. Outbreak.info Research Library: a standardized, searchable platform to discover and explore COVID-19 resources. Nature Methods. Published online February 23, 2023. doi:https://doi.org/10.1038/s41592-023-01770-w
- Grantham R. Amino acid difference formula to help explain protein evolution. Science (New York, NY). 1974;185(4154):862-864. doi:https://doi.org/10.1126/science.185.4154.862
- Daichi Yamasoba, Keiya Uriu, Arnon Plianchaisuk, et al. Virological characteristics of the SARS-CoV-2 omicron XBB.1.16 variant. Published online May 1, 2023. doi:https://doi.org/10.1016/s1473-3099(23)00278-5
- Daichi Yamasoba, Kimura I, Nasser H, et al. Virological characteristics of the SARS-CoV-2 Omicron BA.2 spike. Cell. 2022;185(12):2103-2115.e19. doi:https://doi.org/10.1016/j.cell.2022.04.035
- Kimura I, Yamasoba D, Tamura T, et al. Virological characteristics of the SARS-CoV-2 Omicron BA.2 subvariants, including BA.4 and BA.5. Cell. 2022;185(21):3992-4007.e16. doi:https://doi.org/10.1016/j.cell.2022.09.018
- Tamura T, Ito J, Uriu K, et al. Virological characteristics of the SARS-CoV-2 XBB variant derived from recombination of two Omicron subvariants. Nature Communications. 2023;14(1):2800. doi:https://doi.org/10.1038/s41467-023-38435-3
- Kimura I, Daichi Yamasoba, Nasser H, et al. Multiple mutations of SARS-CoV-2 Omicron BA.2 variant orchestrate its virological characteristics. Journal of virology. 2023;97(10). doi:https://doi.org/10.1128/jvi.01011-23
- Lim H, Baek A, Kim J, et al. Hot spot profiles of SARS-CoV-2 and human ACE2 receptor protein interaction obtained by density functional tight binding fragment molecular orbital method. Scientific Reports. 2020;10(1):16862. doi:https://doi.org/10.1038/s41598-020-73820-8
- Lan J, Ge J, Yu J, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581(215–220). doi:https://doi.org/10.1038/s41586-020-2180-5
- Kaku Y, Okumura K, Padilla-Blanco M, et al. Virological characteristics of the SARS-CoV-2 JN.1 variant. The Lancet Infectious Diseases. 2024;24(2):e82-e82. doi:https://doi.org/10.1016/s1473-3099(23)00813-7
- Jungreis I, Sealfon R, Kellis M. SARS-CoV-2 gene content and COVID-19 mutation impact by comparing 44 Sarbecovirus genomes. Nature Communications. 2021;12(1):2642. doi:https://doi.org/10.1038/s41467-021-22905-7
- Ito J, Suzuki R, Uriu K, et al. Convergent evolution of SARS-CoV-2 Omicron subvariants leading to the emergence of BQ.1.1 variant. Nature Communications. 2023;14(1):2671. doi:https://doi.org/10.1038/s41467-023-38188-z