






In Silico Study of PEX6 Variants in Refsum Disease
In Silico Characterization of Two Adjacent Novel Homozygous Nucleotide Variations in the PEX6 Gene Predict L898V Mutation to Enhance Infantile Refsum Disease and Heimler Syndrome
Lashudaa Madura Madhavan1, Hemavathy Nagarajan2, Umashankar Vetrivel3, Madhavan Jagadeesan1
- Dualhelix Genetic Diagnostics,
- Chennai, Tamil Nadu, India
- Centre for Bioinformatics, KBiRVO, Vision Research Foundation, Chennai, Tamil Nadu, India
- ICMR–Department of Virology and Biotechnology / Bioinformatics Division, National Institute for Research in Tuberculosis, Chennai, Tamil Nadu, India
OPEN ACCESS
PUBLISHED: 30 November 2024
CITATION: Madhavan, L.M., Nagarajan, H., et al., 2024. In Silico Characterization of Two Adjacent Novel Homozygous Nucleotide Variations in the PEX6 Gene Predict L889V Mutation to Enhance Infantile Refsum Disease and Heimler Syndrome. Medical Research Archives, [online] 12(11).
https://doi.org/10.18103/mra.v12i11.0000
COPYRIGHT © 2024 European Society of Medicine. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
DOI https://doi.org/10.18103/mra.v12i11.0000
ISSN 2375-1924
Abstract
Background: Zellweger spectrum disorders are autosomal recessive in origin due to defects in peroxisome biogenesis and with variable severity. The present work aims to characterize a South Asian Indian Zellweger spectrum disorder family with PEX6 mutation for a genotype-phenotype association.
Method: The affected and unaffected individuals in the family were evaluated. A comprehensive examination of the ocular, auditory, dental, integumentary, neuronal, hepato-renal, endocrine, skeletal, cardiac, and other systems was conducted. Investigations deemed fit for diagnosis and management were done. Karyotyping and molecular genetic screening of the peripheral venous blood were performed for aneuploidy and mutation detection. Retinal fundus photograph, optical coherence tomography, bilateral audiogram, magnetic resonance imaging of the brain, ultrasonography of the abdomen, electrocardiography, and echocardiogram were performed. Any non-synonymous nucleotide variations detected were analyzed using in silico methods.
Results: The proband was born of consanguinity and had retinitis pigmentosa in both eyes, bilateral sensorineural deafness, amelogenesis imperfecta, Beau’s line, and punctate leukonychia corresponding to Heimler syndrome. In addition, the proband had developmental nuclear cataracts, ichthyosis, developmental delay, cerebellar ataxia, cognitive deficit, peripheral neuropathy, and muscle movement disorder corresponding to inherited Refsum disease. The proband did not have anosmia. The brain’s magnetic resonance imaging, abdomen ultrasonography, electrocardiography, and echocardiogram were normal. The karyotyping revealed euploidy status. The molecular genetic screening detected two novel adjacent homozygous non-synonymous nucleotide variations c.2691C>A (p.Ser897Arg) and c.2692C>G (p.Leu898Val) in the PEX6 gene. The pathogenic effect, structural destabilization effect, and functional impact of the variants were analyzed through in silico methods. The L898V variant was highly pathogenic compared to the non-conserved S897R variant of PEX6, leading to structural instability and loss of functionality. The molecular dynamics simulation studies also revealed the L898V variant to cause structural instability of PEX6 with higher backbone deviations and residue-wise fluctuations.
Conclusion: This study confirms the significance of L898V variant on the functionality of PEX6 that resulted in a severe disease phenotype. Genetic counseling, followed by multidisciplinary clinical evaluation was required to manage the patient with peroxisomal biogenesis disorder. A phytanic acid-restricted diet may be beneficial to control the infantile Refsum disease severity.
Keywords
Zellweger spectrum disorders, PEX6 mutation, genotype-phenotype association, in silico analysis, infantile Refsum disease, Heimler syndrome.
Introduction
Peroxisomes are single membrane-bound organelles derived from the endoplasmic reticulum. There are several hundred peroxisomes in mammalian cells, each around 1µm in diameter. A single peroxisome carries more than 50 matrix enzymes for various metabolic activities of the mammalian system that include β-oxidation of very long chain fatty acids (VLCFA), α-oxidation of methyl branched phytanic acid, the catabolism, and biosynthesis of ether phospholipids.
Peroxisomal β oxidation utilizes different fatty acid substrates in catabolic and synthetic processes. This includes very long (≥ C22) straight-chain saturated and unsaturated fatty acids (VLCFA), (2 S) methyl-branched chain pristanic acid, and (25 S) di- and tri-hydroxycholestanoic acids (D/THCA). An α-oxidation of methyl-branched phytanic acid is required to convert phytanic acid into pristanic acid.
Chain shortening of VLCFA is the final step in docosahexaenoic acid (DHA) synthesis. A defect in acyl-CoA oxidase 1, D-bifunctional protein, and acetyl-CoA acyltransferase 1 enzymes causes the accumulation of VLCFA to damage the brain, nerve, and adrenals, and a deficiency of DHA affects brain and vision function. β oxidation of methyl-branched chain fatty acid (pristanic acid) and C27 bile acid precursors, D/THCA, with the help of acyl-CoA oxidase 2, D-bifunctional protein, and sterol carrier protein-peroxisomal thiolase results in the chain shortening of pristanic acid. A defect in the enzymes results in the accumulation of pristanic acid, affecting the brain and nerves. An increase in D/THCA causes liver toxicity. The degradation of methyl-branched phytanic acid requires an additional α-oxidation before entering the β-oxidation pathway as pristanic acid. A defect in the enzyme phytanyl-CoA hydroxylase (PHYH) causes tissue accumulation of phytanic acid, which results in retinal degeneration, cerebellar degeneration, and peripheral neuropathy.
Nuclear genes code for the proteins found in the membrane and the lumen of peroxisomes. Proteins named ‘peroxins’ deliver proteins from the cytoplasm to the peroxisomal membrane and into the lumen. More than 20 peroxins participate in the biogenesis and maintenance of peroxisomes. A defect in any of the peroxins due to mutation causes deregulation of peroxisomal biogenesis. This critically affects the metabolic pathways that result in the accumulation of by-products and cause damage to ocular, auditory, dental, integumentary, neuronal, hepato-renal, endocrine, skeletal, cardiac, growth, and intellectual capabilities.
The peroxisome biogenesis disorder (PBD) represents a pattern of metabolic malformation syndromes. Biallelic pathogenic mutation in any of the fourteen PEX genes causes peroxisomal assembly disorders. Defects in PEX1, PEX2, PEX3, PEX5, PEX6, PEX10, PEX11β, PEX12, PEX13, PEX14, PEX16, PEX19, PEX26, and PEX7 genes cause Zellweger syndrome disorder (ZSD) and Rhizomelic Chondrodysplasia Punctata type I (RCDP1) respectively. The severity of the disease correlates with the age of onset of the symptoms, peroxisome number, the impact of the mutation on peroxin function, and residual enzyme function. The overall birth incidence of ZSD is 1 in 50,000 and 1 in 100,000 for RCDP1.
ZSD has variable severity, which includes 1. Zellweger Syndrome (ZS) due to PEX1 mutation results in a severe form of the disease characterized by craniofacial, neurological, and multi-organ dysfunction noticed in the first year of life with high mortality. 2. X-linked Neonatal Adrenoleukodystrophy (NALD) due to ABCD1 mutation is an intermediate form of the disease characterized by muscle hypotonia, severe psychomotor retardation, and failure to thrive. It may also lead to blindness and deafness. 3. Infantile Refsum Disease (IRD) due to PEX6 mutation is milder, and 4. Heimler syndrome (HS) due to PEX1 and PEX6 is a milder form of ZSD. RCDP1, due to a PEX7 gene defect characterized by skeletal abnormalities, distinctive facial features, intellectual abnormality, and respiratory problems.
In the present study, the clinical features of HS and IRD observed in the proband due to PEX6 mutations were subjected to a genotype-phenotype association to understand the disease severity in a 24-year-old female.
Methods
CLINICAL EVALUATION
The patient and the family members were recruited for the study after obtaining written informed consent, and the guidelines of the Declaration of Helsinki were followed.
A physical examination of the proband and her parents was performed. A comprehensive assessment of the ocular, auditory, dental, integumentary, neuronal, hepato-renal, endocrine, skeletal, cardiac, and other systems was conducted. The ophthalmic evaluation included visual acuity for distance and near (Snellen’s visual acuity chart), color vision (Ishihara pseudo isochromatic color vision plates), refraction, intraocular pressure (IOP) (Applanation Tonometer, Topcon Medical Systems, NJ, USA), clinical biomicroscopy, and a dilated fundus examination using slit lamp/90D and indirect ophthalmoscopic examination. The proband’s birth, development, and family history were obtained.
The family underwent molecular characterization of the disease. Five millilitres of peripheral venous blood were collected, and DNA extraction was performed using a Qiagen DNA kit (Qiagen, India). The proband DNA sample was subjected to a clinical exome panel using next-generation sequencing technology for mutation detection (Medgenome Labs, India). Any changes observed were confirmed bidirectionally through Sanger’s sequencing technology. The pathogenicity of novel variation was confirmed through family segregation analysis and screening of 100 normal control chromosomes. The pathogenic effect, structural destabilization effect, and functional impact of the variants were evaluated through in silico methods.
IN SILICO ANALYSIS OF THE VARIANTS
The variants of PEX6 were first investigated through conservation analysis using Consurf to determine the evolutionary conservation of amino acids and predict their functional relevance. Following this, all variants underwent a sequence-based analysis using PredictSNP which assesses the deleterious effect of the variants. Additionally, the functional impact of the variants was analyzed using MutPred. A sequence-based analysis was initially conducted due to the lack of a three-dimensional crystal structure for PEX6. The PEX6 protein sequence (UniProt ID: Q13608) was retrieved, and its regions of intrinsic disorder were predicted using the D2P2 server. Additionally, the protein’s secondary structure elements were predicted with Psipred to gain insight into the possible arrangement of helices, sheets, and loops within the protein. The AlphaFold model of PEX6 (AF-Q13608-F1) was employed for further structural destabilization analysis. The disordered segments identified in the earlier analysis were removed from the model. The Schrödinger Protein Preparation Wizard was engaged to pre-process and optimize the structural model for subsequent stability evaluations. To evaluate the effect of the variants on the structural stability of PEX6, several in silico tools namely, I-Mutant 2.0, DynaMut, DUET, SDM, mCSM were used. Based on the comprehensive analysis, only variants predicted to be pathogenic, destabilizing, and affecting functionality were selected for further structural modeling. These selected variants were modeled using the PyMOL tool, and the models were further refined using the Schrödinger suite. To further evaluate the stability and conformational variations of both the wild type (WT) and the selected pathogenic variants, molecular dynamics (MD) simulations were conducted using GROMACS. MD simulations provide a dynamic view of the protein’s behavior in a simulated environment, capturing details about stability, flexibility, and conformational changes that might result from specific mutations. These simulations help to understand how the variants affect the protein’s overall dynamics compared to the WT.
Results
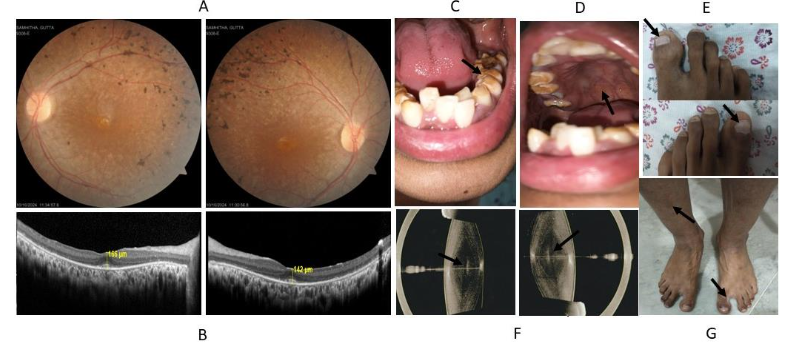
The twenty-four-year-old female was evaluated for side and night vision defects noticed at five years of age. Her best corrected visual acuity was 6/6+2 in the right eye and 6/9+3 in the left eye. Her refractive error correction was RE: +/- Dsph with -2.50 Dcyl at 40° & LE: +/- Dsph with -2.50 Dcyl at 140°. The intraocular pressure in both eyes was 16 mm Hg. Anterior segment examination revealed a developmental nuclear cataract in both eyes. Posterior segment examination revealed a normal optic disc, while the macula showed early degeneration in both eyes. There was a narrowing of blood vessels with bony spicules along the peripheral retina suggestive of retinitis pigmentosa. There was severe retinal pigment epithelial (RPE) mottling throughout the retina. Optical coherence tomography revealed a thinned-out outer nuclear (photoreceptor layer) and outer plexiform layers. Severe retinal pigment epithelial hyperplasia was noted along the peripheral retina in both eyes. After refractive error correction, the parents’ visual acuity was normal in both eyes. The parents had nuclear cataracts in both eyes and their intraocular pressure was normal. The posterior segment examinations of the parents were normal.
Physical examination of the other systems of the proband revealed bilateral sensorineural hearing impairment, amelogenesis imperfecta, Beau’s line, and punctate leukonychia corresponding to HS. In addition to the above abnormality, the proband had developmental nuclear cataracts in both eyes, ichthyosis, developmental delay, cerebellar ataxia, cognitive deficit, peripheral neuropathy, and muscle movement disorder corresponding to IRD. The proband did not have anosmia. Hallux varus deformity was noted. The brain’s magnetic resonance imaging, abdomen ultrasonography, electrocardiography, and echocardiogram were normal. The karyotyping revealed euploidy status.
The proband was born through lower segment cesarean section due to premature rupture of the membrane a week before parturition. The birth weight was normal as per Asian-Indian standards. She cried immediately after birth; her sucking reflex was normal. Her early milestones of a social smile, head holding, sitting with and without support, and standing with support were normal. Her walking was delayed by 5 months. She had a delay in speaking. Her teeth started to erupt on time but were irregularly arranged and discolored with enamel loss. She had bilateral sensorineural hearing impairment and a side vision defect in both eyes noticed at five years of age. At the age of ten, her intelligence was below normal. She had difficulty understanding, writing, memorizing, reciprocating, counting, and expressing. After puberty, she had episodes of mood swings, bipolar disorder, and delusion. The proband was born through consanguinity, and her parents were normal.
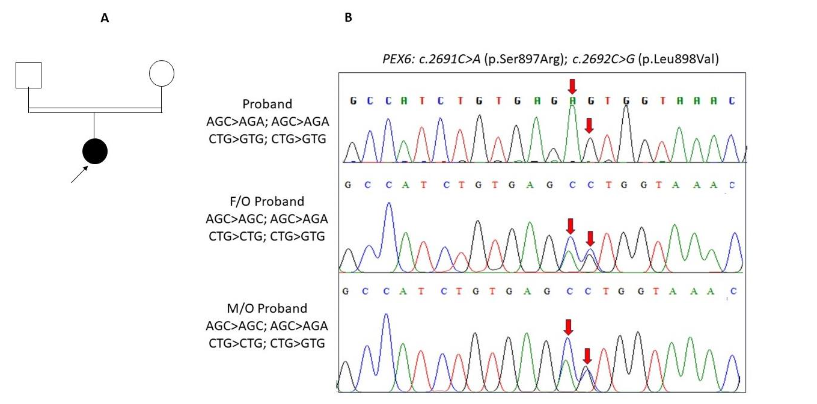
The molecular genetic screening detected two novel adjacent homozygous non-synonymous nucleotide variations c.2691C>A (p.Ser897Arg) and c.2692C>G (p.Leu898Val) in the PEX6 gene. In silico analysis of the variants was assessed to understand the severity of the variation and its impact on the disease severity.
IN SILICO ANALYSIS OF THE VARIANTS
The evolutionary analysis of PEX6 provided valuable insights into the significance of specific residues. Among the variants studied, residue S897 was identified as the least conserved and surface-exposed, suggesting that mutations at this position may have minimal functional significance. Conversely, L898 was highly conserved, indicating its critical role in PEX6’s structure and function. Therefore, any mutation at position L898 would impact the structure or functionality of PEX6.
Pathogenicity predictions further supported this conclusion. In pathogenicity analysis, the L898V variant was classified as highly pathogenic, with three predictors yielding high accuracy scores (above 0.68), indicating its deleterious potential. In contrast, the S897R variant showed the least pathogenic effect, with lower scores from the pathogenicity predictors. Despite the least deleterious effect of S897R, both the variants were subjected to functionality impact analysis, and it was observed that the L898V variant had a high MutPred score of 0.629, suggesting significant functional disruptions, including a loss of solvent accessibility, and altered metal binding properties. In contrast, the S897R variant had a lower score of 0.369, suggesting minimal functional impact. Since S897R was found non-deleterious with minimal functional implications, only the L898V variant underwent further structural stability analysis. Before the structural analysis, the disorderliness of PEX6 was observed at the initial N-terminal ranging as follows: 57-72, 167-182, 236-237, 257-332, and these segments were observed to be highly composed of loop (high confidence) and helices (low confidence). Only the structured segment of Alpha fold PEX6 (344-980) refined structure was processed for structural stability analysis. The results indicated that the L898V variant significantly destabilizes the structure of PEX6. This finding underscores the critical role of the L898 residue in maintaining the structural integrity and functionality of the PEX6 protein, while the S897 variant appears to have minimal effects.
| Variant information | Wild residue | Position | Target residue | Pathogenicity predictors |
|---|---|---|---|---|
| S | L | 897 | 898 | PredictSNP NEUTRAL, MAPP NEUTRAL, PhD-SNP DELETERIOUS, PolyPhen-1 NEUTRAL, PolyPhen-2 NEUTRAL, SIFT NEUTRAL, SNAP DELETERIOUS |
| VARIANT | I-Mutant2.0 ΔΔG | mCSM (kcal/mol) ΔΔG | SDM (kcal/mol) ΔΔG | DUET (kcal/mol) ΔΔG | Dynamut (kcal/mol) |
|---|---|---|---|---|---|
| L898V | Decrease -1.040 (Destabilizing) | -0.610 (Destabilizing) | -0.845 (Destabilizing) | -0.384 (Destabilizing) |
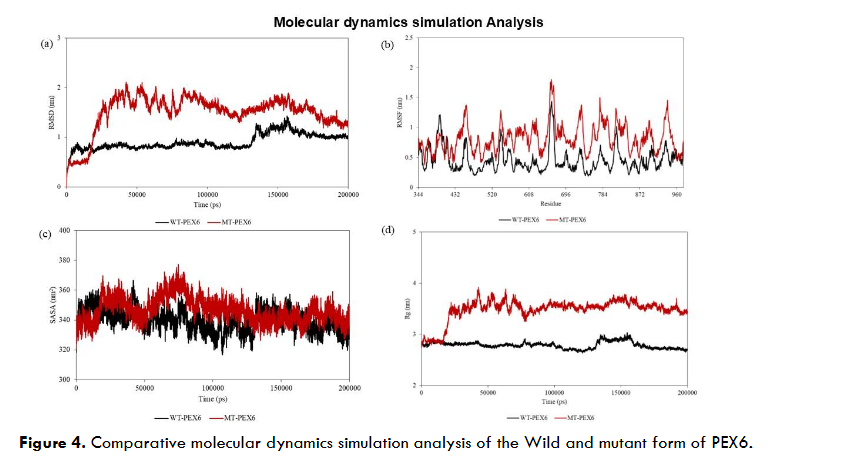
Furthermore, the molecular dynamics (MD) simulation analysis comparing the wild-type (WT) and mutant (MT-L898V) forms of PEX6 revealed distinct differences in their stability and flexibility. The root mean square deviation (RMSD) analysis showed that the MT-PEX6 exhibited higher RMS deviations (~1.98-2 nm) compared to the WT-PEX6. The WT-PEX6 initially demonstrated a stable equilibrium with an RMSD of ~1 nm until about 130 ns, after which a sudden rise occurred, reaching an RMSD of ~1.4 nm at around 140 ns. Despite this, the WT-PEX6 attempted to converge during the final ~30 ns of the MD run. In contrast, MT-PEX6 displayed higher RMS deviations early in the simulation, reaching ~2 nm up to 90 ns. After ~120 ns, the MT-PEX6 system showed signs of equilibration, maintaining an RMSD of ~1.8 nm for the remainder of the simulation. The root mean square fluctuation (RMSF) profile indicated that MT-PEX6 residues exhibited significantly higher fluctuations (ranging from ~0.8-1.8 nm) compared to WT-PEX6, where the maximum fluctuation was ~1.4 nm. Notably, the segment between residues 661-664 showed substantial fluctuation in MT-PEX6. Furthermore, the L898V mutant residue exhibited higher fluctuations in the MT-PEX6, with an RMSF value of 0.85 nm, in contrast to 0.45 nm in the WT-PEX6. The radius of gyration (Rg) analysis indicated that while both the WT and MT forms of PEX6 exhibited minimal changes in compactness, the average Rg value for WT-PEX6 was 2.790 nm and the MT-PEX6 had a slightly higher average Rg value of 3.481 nm. These results suggest that the MT form has a looser structure, although the difference in compactness was negligible. Additionally, solvent-accessible surface area (SASA) analysis revealed that the WT-PEX6 had an average SASA value of 339.808 nm², while the MT-PEX6 exhibited a slightly higher value of 346.645 nm². This increase in SASA suggests that the MT form of PEX6 may have a marginally more exposed surface, potentially affecting interactions with other molecules.
Discussion
Peroxisomes are simple organelles of a single membrane that forms a lumen containing many oxidative enzymes. Two types of targeting signals direct proteins from the cytoplasm to the peroxisome lumen. The PEX5 imports carrier proteins with the help of type 1 peroxisomal targeting signal (PTS1) to the peroxisomal lumen. The PEX5 soluble protein is proposed to bind with the PTS1 motif and insert into the lipid bilayer surrounding the peroxisomes, where PEX5 forms a transient pore to deliver the cargo protein into the lumen. After the delivery, the PEX5 returns to the cytoplasm for further rounds of imports. The PEX5 is recycled in conjugation with ubiquitin and ATP hydrolysis by a AAA ATPase with the help of PEX6, which acts as a recycling matrix import machinery. Apart from PEX6, the recycling matrix import delivery is supported by PEX1, PEX15, PEX26, PEX2, PEX4, PEX10, PEX12, and PEX22. A similar mode of action is proposed for PEX7, the import receptor for type 2 peroxisomal targeting signal (PTS2) proteins—defect in PEX6 governing the peroxisome assembly results in a decrease in PEX5-PTS1 complex’s activity. This causes a decreased catalytic activity towards VLCFA and decreases the synthesis of DHA. The tissue accumulation of VLCFA causes brain, nerve, and adrenal damage—the deficiency of DHA results in brain damage and a decrease in vision.
There was a concordance in most of the characteristic features observed in this study with the classical features reported earlier for Infantile Refsum disease and Heimler syndrome. At the same time, there was a discordant in some of the features, which was reported as classical for HS and IRD. The nail features observed in HS in earlier studies weren’t noticed with the same severity. Similarly, the feature of anosmia noticed in IRD by previous studies was not found in the proband. Further, amelogenesis imperfecta is associated with a defect in three major genes (AMELX, ENAM, and MMP20) responsible for teeth, and enamel development. As the disease causal variation was not detected in these genes in the present study, it provides a space to investigate the role of PEX6 in the development of normal teeth. As the number of patients identified with PBD is less, identifying and documenting more patients with PBD will give a better understanding of the condition, which may help to decipher the molecular wiring of this disorder and may offer a potential treatment such as gene replacement and stem cell-based therapies in coming years.
The present study assessed the impact of the two novel adjacent homozygous non-synonymous nucleotide variations, S897R and L898V, in the PEX6 for a genotype-phenotype correlation. After the phenotype characterization, the two novel variations were assessed for a possible severity prediction. The multi-step in silico analysis of PEX6 variants implemented in this study provides comprehensive insights into how specific mutations impact the protein’s function and structure. By integrating evolutionary conservation, sequence-based analysis, structural stability, and functional impact assessments, the study elucidates the pathogenicity of variants such as L898V and S897R. Evolutionary analysis revealed that residue S897 is surface-exposed and the least conserved, suggesting that mutations like S897R would have minimal effects on function. On the other hand, L898 is highly conserved, indicating its crucial role in PEX6 structure and function. The L898V variant was predicted to be highly pathogenic across multiple sequence-based and structural prediction tools, whereas S897R had minimal pathogenic effects. Functional impact analysis using MutPred showed that L898V significantly affects PEX6 functions, such as solvent accessibility and metal binding, while S897R had little effect. Structural stability analysis confirmed that the L898V mutation destabilizes the PEX6 protein, underscoring L898’s importance in maintaining structural integrity. Molecular dynamics simulations further highlighted the destabilizing effects of the L898V variant. The L898V mutation in PEX6 leads to greater structural instability, increased residue fluctuations, and a slightly more exposed conformation, as indicated by the RMSD, RMSF, Rg, and SASA analyses. These changes suggest that the mutation may impact the functional dynamics of the protein. In conclusion, the L898V mutation in PEX6 leads to significant structural instability and altered protein dynamics, suggesting its potential role in PEX6-related diseases.
Conclusion
A multidisciplinary approach is required to manage patients with peroxisomal biogenesis disorder. Genetic counseling was offered to the family with a focus on PEX6 mutation on the nature of the disease, the risk of transmission to the subsequent generations, family segregation analysis, and possible future treatment modalities such as gene replacement therapy and stem cell-based therapies were discussed. The application of low vision aids, cochlear implants, dentition implants, skin care, regular cardiac assessment, and management of peripheral neuropathy and muscle movement disorders were emphasized. Rehabilitation and regular follow-up were insisted. The patient was advised to reduce the intake of a phytanic acid-rich diet such as red meat, dairy products, and fatty fish. A regular check on the phytanic acid level in the blood was advised. The role of plasmapheresis in extremely high-level phytanic acid was also discussed.
Conflicts of Interest
There is no conflict of interest among the authors.
Funding
None
Acknowledgment
Our sincere gratitude to the patient and the family for participating in the study.
References
- W.E. Bowers, Christian de Duve and the discovery of lysosomes and peroxisomes. Trends Cell Biol. 1998; 8:330-333. doi: 10.1016/s0962-8924(98)01314-2
- Wanders RJ, Waterham HR. Biochemistry of mammalian peroxisomes revisited. Annu Rev Biochem. 2006; 75:295-332. doi: 10.1146/annurev.biochem.74.082803.133329
- Argyriou C, D’Agostino MD, Braverman N. Peroxisome biogenesis disorders. Transl Sci Rare Dis. 2016; 1:111-144. doi: 10.3233/TRD-160003
- Ferdinandusse S, Denis S, Dacremont G, Wanders R. Studies on the metabolic fate of n-3 polyunsaturated fatty acids. J Lipid Res. 2003; 44:1992–1997.
- Moore SA, Hurt E, Yoder E, Sprecher H, Spector AA. Docosahexaenoic acid synthesis in human skin fibroblasts involves peroxisomal retroconversion of tetracosahexaenoic acid, J Lipid Res. 1995; 36:2433–2443.
- Wanders RJ, van Roermund CW, van Wijland MJ, Heikoop J, Schutgens RB, Schram AW, Tager JM, van den Bosch H, Poll-Thé BT, Saudubray JM. Peroxisomal very long-chain fatty acid beta-oxidation in human skin fibroblasts: activity in Zellweger syndrome and other peroxisomal disorders. Clin Chim Acta. 1987; 166:255-63. doi: 10.1016/00098981(87)90428-1
- Singh H and Poulos A. Distinct long chain and very long chain fatty acyl CoA synthetases in rat liver peroxisomes and microsomes. Arch Biochem Biophys. 1988; 266:486–495.
- Street JM, Johnson DW, Singh H, Poulos A. Metabolism of saturated and polyunsaturated fatty acids by normal and Zellweger syndrome skin fibroblasts. Biochem J. 1989; 15; 260:647-55. doi: 10.1042/bj2600647.
- Uchiyama A, Aoyama T, Kamijo K, Uchida Y, Kondo N. Orii T. Molecular cloning of cDNA encoding rat very long-chain acyl-CoA synthetase. J Biol Chem. 1996; 271:30360–30365.
- Schepers L, VanVeldhoven PP, Casteels M, Eyssen HJ, Mannaerts GP. Presence of three acyl-CoA oxidases in rat liver peroxisomes, An inducible fatty acyl-CoA oxidase, a noninducible fatty acyl-CoA oxidase, and a noninducible trihydroxycoprostanoyl-CoA oxidase. J Biol Chem. 1990; 265:5242–5246.
- Mize CE, Steinberg D, Avigan J and Fales HM. A pathway for oxidative degradation of phytanic acid in mammals. Biochem Biophys Res Commun. 1966; 25:359–365.
- Eldjarn L, Heredopathia atactica polyneuritiformis (Refsum’s disease)–a defect in the omega-oxidation mechanism of fatty acids. Scand J Clin Lab Invest. 1965; 17:178–181.
- Schrader M, Thiemann M, Fahimi HD. Peroxisomal motility and interaction with microtubules. Microsc Res Tech. 2003; 61:171-8. doi: 10.1002/jemt.10326
- Islinger M, Voelkl A, Fahimi HD, Schrader M. The peroxisome: an update on mysteries 2.0. Histochem Cell Biol. 2018; 150:443-471. doi: 10.1007/s00418-018-1722-5
- Kumar R, Islinger M, Worthy H, Carmichael R, Schrader M. The peroxisome: an update on mysteries 3.0. Histochem Cell Biol. 2024; 16:99-132. doi: 10.1007/s00418-023-02259-5
- Gould SJ, Raymond GV and Valle D, The peroxisome biogenesis disorders, In: The Metabolic and Molecular Bases of Inherited Disease. McGraw Hill International Book Company, New York, NY. 2001; 3181–3217.
- Stoll C, Dott B, Roth MP and Alembik Y. Birth prevalence rates of skeletal dysplasias, Clin Genet. 1989; 35:88–92.
- Klouwer FC, Berendse K, Ferdinandusse S, Wanders RJ, Engelen M, Poll-The BT. Zellweger spectrum disorders: clinical overview and management approach. Orphanet J Rare Dis. 2015; 10:151. doi: 10.1186/s13023-015-0368-9
- Goldfischer S, Moore CL, Johnson AB, Spiro AJ, Valsamis MP, Wisniewski HK. Peroxisomal and mitochondrial defects in the cerebro-hepato-renal syndrome, Science. 1973; 182:62-64. doi: 10.1126/science.182.4107.62
- Kelley RI, Datta NS, Dobyns WB, Hajra AK, Moser AB, Noetzel MJ. Neonatal adrenoleukodystrophy: New cases, biochemical studies, and differentiation from Zellweger and related peroxisomal polydystrophy syndromes, Am J Med Genet. 1986; 23:869-901. doi: 10.1002/ajmg.1320230404
- Vamecq J, Draye JP, Van Hoof F, Misson JP, Evrard P, Verellen G, Eyssen HJ, Van Eldere J, Schutgens RB, Wanders RJ. Multiple peroxisomal enzymatic deficiency disorders. A comparative biochemical and morphologic study of Zellweger cerebrohepatorenal syndrome and neonatal adrenoleukodystrophy. Am J Pathol; 1986; 125:524-35.
- Lee PR, Raymond GV. Child neurology: Zellweger syndrome. Neurology. 2013; 14:80:e207-10. doi: 10.1212/WNL.0b013e3182929f8e
- Bose M, Yergeau C, D’Souza Y, Cuthbertson DD, Lopez MJ, Smolen AK, Braverman NE. Characterization of Severity in Zellweger Spectrum Disorder by Clinical Findings: A Scoping Review, Meta-Analysis and Medical Chart Review. Cells. 2022; 11:1891. doi: 10.3390/cells11121891
- Powers JM, Moser HW. Peroxisomal disorders: genotype, phenotype, major neuropathologic lesions, and pathogenesis. Brain Pathol. 1998; 8:101-20. doi: 10.1111/j.1750-3639
- Corzo D, Gibson W, Johnson K, Mitchell G, LePage G, Cox GF, Casey R, Zeiss C, Tyson H, Cutting GR, Raymond GV, Smith KD, Watkins PA, Moser AB, Moser HW, Steinberg SJ. Contiguous deletion of the X-linked adrenoleukodystrophy gene (ABCD1) and DXS1357E: a novel neonatal phenotype similar to peroxisomal biogenesis disorders. Am J Hum Genet. 2002; 70:1520-31. doi: 10.1086/340849
- Poll-The BT, Saudubray JM, Ogier H, Schutgens RB, Wanders RJ, Schrakamp G, van den Bosch H, Trijbels JM, Poulos A, Moser HW, et al. Infantile Refsum’s disease: biochemical findings suggesting multiple peroxisomal dysfunction. J Inherit Metab Dis. 1986; 9:169-74. doi: 10.1007/BF01799455
- Naidu S, Moser H. Infantile Refsum disease. Am J Neuroradiol. 1991; 12:1161-3.
- Slanina AM, Coman AE, Anton-Păduraru DT, Popa E, Barbacariu CL, Novac O, Petroaie AD, Bacușcă AI, Manole M, Cosmescu A. PEX6 Mutation in a Child with Infantile Refsum Disease-A Case Report and Literature Review. Children (Basel) 2023; 10:530. doi: 10.3390/children10030530
- Ratbi I, Falkenberg KD, Sommen M, Al-Sheqaih N, Guaoua S, Vandeweyer G, Urquhart JE, Chandler KE, Williams SG, Roberts NA, El Alloussi M, Black GC, Ferdinandusse S, Ramdi H, Heimler A, Fryer A, Lynch SA, Cooper N, Ong KR, Smith CE, Inglehearn CF, Mighell AJ, Elcock C, Poulter JA, Tischkowitz M, Davies SJ, Sefiani A, Mironov AA, Newman WG, Waterham HR, Van Camp G. Heimler Syndrome Is Caused by Hypomorphic Mutations in the Peroxisome-Biogenesis Genes PEX1 and PEX6. Am J Hum Genet. 2015; 97:535-45. doi: 10.1016/j.ajhg.2015.08.011
- Smith CE, Poulter JA, Levin AV, Capasso JE, Price S, Ben-Yosef T, Sharony R, Newman WG, Shore RC, Brookes SJ, Mighell AJ, Inglehearn CF. Spectrum of PEX1 and PEX6 variants in Heimler syndrome. Eur J Hum Genet. 2016; 24:1565-1571. doi: 10.1038/ejhg.2016.62
- Heimler A, Fox JE, Hershey JE, Crespi P: Sensorineural hearing loss, enamel hypoplasia, and nail abnormalities in sibs. Am J Med Genet. 1991; 39:192-195. doi: 10.1002/ajmg.1320390214
- Neuhaus C, Eisenberger T, Decker C, Nagl S, Blank C, Pfister M, Kennerknecht I, Müller-Hofstede C, Charbel Issa P, Heller R, Beck B, Rüther K, Mitter D, Rohrschneider K, Steinhauer U, Korbmacher HM, Huhle D, Elsayed SM, Taha HM, Baig SM, Stöhr H, Moeller F, Markus S. Next-generation sequencing reveals the mutational landscape of clinically diagnosed Usher syndrome: copy number variations, phenocopies, a predominant target for translational read-through, and PEX26 mutated in Heimler syndrome. Mol Genet Genomic Med. 2017; 5:531-552. doi: 10.1002/mgg3.312
- Daich Varela M, Jani P, Zein WM, D’Souza P, Wolfe L, Chisholm J, Zalewski C, Adams D, Warner BM, Huryn LA, Hufnagel RB. The peroxisomal disorder spectrum and Heimler syndrome: Deep phenotyping and review of the literature. Am J Med Genet C Semin Med Genet. 2020; 184:618-630. doi: 10.1002/ajmg.c.31823
- Braverman N, Steel G, Obie C, Moser A, Moser H and Gould S.J. Human PEX7 encodes the peroxisomal PTS2 receptor and is responsible for rhizomelic chondrodysplasia punctata. Nat Genet.1997; 15:369-376. doi: 10.1038/ng0497-369
- Jacobsen JC, Glamuzina E, Taylor J, Swan B, Handisides S, Wilson C, Fietz M, van Dijk T, Appelhof B, Hill R, Marks R, Love DR, Robertson SP, Snell RG, Lehnert K. Whole Exome Sequencing Reveals Compound Heterozygosity for Ethnically Distinct PEX7 Mutations Responsible for Rhizomelic Chondrodysplasia Punctata, Type 1. Case Rep Genet. 2015; 2015:454526. doi: 10.1155/2015/454526
- Fallatah W, Cui W, Di Pietro E, Carter GT, Pounder B, Dorninger F, Pifl C, Moser AB, Berger J, Braverman NE. A Pex7 Deficient Mouse Series Correlates Biochemical and Neurobehavioral Markers to Genotype Severity-Implications for the Disease Spectrum of Rhizomelic Chondrodysplasia Punctata Type 1. Front Cell Dev Biol. 2022 11;10:886316. doi: 10.3389/fcell.2022.886316
- Chorin AB , Masrati G , Kessel A , Narunsky A , Sprinzak J, Lahav S, Ashkenazy H. ConSurf-DB: An accessible repository for the evolutionary conservation patterns of the majority of PDB proteins. Protein Sci. 2020; 29:258-267. doi: 10.1002/pro.3779
- Bendl J, Stourac J, Salanda O , Pavelka A, Wieben ED, Zendulka J, Brezovsky J, Damborsky J. PredictSNP: robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput Biol. 2014; 10:e1003440. doi: 10.1371/journal.pcbi.1003440
- Pejaver P, Urresti J, Lugo-Martinez J, Pagel AK, Lin GN, Nam JU, Mort M, Cooper DN, Sebat J, Iakoucheva LM, Mooney SD. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat Commun. 2020; 20:11:5918. doi: 10.1038/s41467-020-19669-x
- Oates ME , Romero P, Ishida T, Ghalwash M, Mizianty MJ, Xue B, Dosztányi Z, Uversky VN, Obradovic Z, Kurgan L, Dunker AK, Gough J. D²P²: database of disordered protein predictions. Nucleic Acids Res. 2013; 41:508-516. doi: 10.1093/nar/gks1226
- Buchan DWA, Jones DT. The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res. 2019; 47:402-407. doi: 10.1093/nar/gkz297
- Sastry GM, Adzhigirey M, Day T, Ramakrishna Annabhimoju, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013; 27:221-34. doi: 10.1007/s10822-013-9644-8
- Capriotti E, Fariselli P, Casadio R. I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005; 33:306-310. doi: 10.1093/nar/gki375.
- Rodrigues CH. , Pires DE, Ascher DB. DynaMut: predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 2018; 46:350-355. doi: 10.1093/nar/gky300
- Pires DE, Ascher DB, Blundell TL. DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 2014; 42:W314-9. doi: 10.1093/nar/gku411.
- Worth CL, Preissner R, Blundell TL. SDM–a server for predicting effects of mutations on protein stability and malfunction. Nucleic Acids Res. 2011; 39:215-22. doi: 10.1093/nar/gkr363
- Pires DEV, Ascher DB, Blundell TL. mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics. 2014; 30:335-42. doi: 10.1093/bioinformatics/btt691
- Kavitha B, Ranganathan S, Gopi S, Vetrivel U, Hemavathy N, Mohan V, Radha V. Molecular characterization and re-interpretation of HNF1A variants identified in Indian MODY subjects towards precision medicine. Front Endocrinol (Lausanne). 2023; 16;14:1177268. doi: 10.3389/fendo.2023.1177268.
- H.W. Platta, S. Grunau, K. Rosenkranz, W. Girzalsky and R. Erdmann. Functional role of the AAA peroxins in dislocation of the cycling PTS1 receptor back to the cytosol. Nat Cell Biol. 2005; 7:817–822. doi: 10.1038/ncb1281
- D. Tan, N.B. Blok, T.A. Rapoport and T. Walz, Structures of the double-ring AAA ATPase Pex1-Pex6 involved in peroxisome biogenesis. FEBS J. 2016; 283:986–992. doi: 10.1111/febs.13569
- Sá MJ, Rocha JC, Almeida MF, Carmona C, Martins E, Miranda V, Coutinho M, Ferreira R, Pacheco S, Laranjeira F, Ribeiro I, Fortuna AM, Lacerda L. Infantile Refsum Disease: Influence of Dietary Treatment on Plasma Phytanic Acid Levels. JIMD Rep. 2016; 26:53-60. doi: 10.1007/8904_2015_487