Iron Deficiency and Cancer: Mechanisms and Risks
Iron and Micronutrient Deficiencies as Drivers of Carcinogenesis and Therapeutic Resistance in Cancer: An In-Depth Clinical and Physiological Three-Part Review
Part I—Iron Deficiency, Insufficiency, Excess, and Neoplasia
Glenn Tisman, M.D.1
- Retired Medical Oncologist, Independent Researcher, formerly Associate in Pathology, Columbia University; Fellow in Medicine-Hematology-Oncology, Bronx VA/Mount Sinai/Mt. Sinai Hospital Joint Program; Fellow in Medical Oncology, Keck School of Medicine, USC
OPEN ACCESS
PUBLISHED: 31 December 2025
CITATION: Tisman, G., 2025. Iron and Micronutrient Deficiencies as Drivers of Carcinogenesis and Therapeutic Resistance in Cancer: An In-Depth Clinical and Physiological Three-Part Review (Part I—Iron Deficiency, Insufficiency, Excess, and Neoplasia). Medical Research Archives, [online] 13(12). https://doi.org/10.18103/mra.v13i12.7182
COPYRIGHT: © 2025 European Society of Medicine. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
DOI https://doi.org/10.18103/mra.v13i12.7182
ISSN 2375-1924
ABSTRACT
The 2019 Nobel Prize in Physiology or Medicine underscored a fundamental insight: cellular oxygen-sensing and hypoxia pathways orchestrate key biological responses, including the initiation and progression of cancer. Building on this framework, this review translates the core science of oxidative stress and hypoxia-driven molecular alterations into practical clinical strategies. Latent iron deficiency and iron overload are both common, frequently underdiagnosed, and potent mediators of redox activity capable of driving carcinogenesis. This manuscript presents a mechanistic narrative review of how iron deficiency and iron overload generate reactive oxygen species (ROS), promote tumor initiation, progression, and resistance to therapy. Emphasis is placed on equipping clinicians who manage patients with hereditary cancer predisposition, subclinical premalignant lesions, or early biomarkers of oncogenesis to recognize iron imbalance earlier and implement targeted surveillance and timely intervention before developing neoplastic lesions.
Keywords
Iron deficiency, micronutrient deficiencies, carcinogenesis, therapeutic resistance, oxidative stress, hypoxia.
Introduction
Part I of this three-manuscript series examines the fundamental pathophysiology of oxidative stress and its role in oncogenesis. In this installment, we focus on how imbalances in essential micronutrients exacerbate oxidative stress and how cellular hypoxia–driven signaling pathways both initiate and sustain pro-carcinogenic processes, often progressing silently below the threshold of routine clinical detection. Attention to this issue was first drawn in 1992 by Dr Victor Herbert, who asserted, “Everyone Should be Tested for Iron Disorders.” In 2018, insights from Dr Esa Soppi of Finland reinforced the enduring importance of early detection and management of subclinical iron deficiency, a potent source of oxidative stress, as well as deficiencies of other micronutrients and their contribution to prolonged, clinically undetected neoplasia.
Aim and scope of Part I
In this first installment, we focus on how iron insufficiency and iron overload perturb redox balance and oxygen-sensing pathways, particularly reactive oxygen species (ROS), hypoxia-inducible factors (HIF-1 and HIF-2), nuclear factor κB (NF-κB), and nuclear factor erythroid 2–2-related factor 2 (Nrf2), to promote tumor initiation, progression, and resistance to therapy. This is a mechanistic narrative review centered on adult human malignancies in which iron imbalance is plausibly involved in carcinogenesis or treatment response; it synthesizes experimental, translational, and selected clinical data rather than providing a systematic trial meta-analysis. Other micronutrient deficiencies (folate, vitamin B12, vitamin B6, and vitamin D) and their epigenetic consequences are introduced only when they directly interact with iron-related pathways and will be developed in detail in Parts II and III.
Iron Metabolism
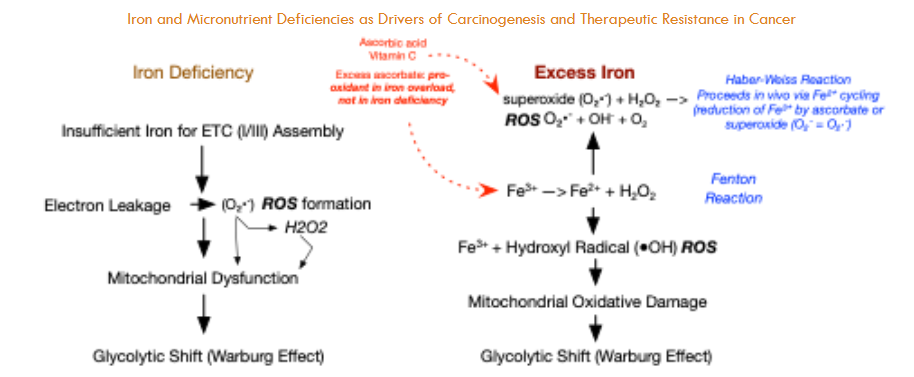
Disruptions in iron metabolism, whether due to deficiency or overload, impair cellular energy production and oxygen handling, leading to the accumulation of reactive oxygen species (ROS). These unstable oxygen-containing molecules damage DNA, proteins, and cell membranes.
In iron deficiency, mitochondrial respiration is compromised, causing electron-transport chain leakage and excess ROS generation. Conversely, iron overload promotes ROS formation via the Fenton reaction, in which Fe²⁺ catalyzes hydrogen peroxide into highly reactive hydroxyl radicals. Both scenarios culminate in oxidative stress, driving tissue injury, reducing therapeutic efficacy, and contributing to the progression of numerous diseases.
Hypoxia-Inducible Factors (HIFs)
Cells depend on hypoxia-inducible factors (HIFs) to gauge oxygen levels and adjust gene expression, which may foster carcinogenesis. HIF-1α and HIF-2α are protein transcription factors that act as cellular “oxygen gauges.” Under normal oxygen conditions, and with sufficient iron, HIF-α subunits are hydroxylated by prolyl hydroxylases (iron-dependent enzymes) and rapidly degraded, keeping intracellular HIF levels very low. When oxygen availability falls or iron-dependent degradation is impaired (for example, due to iron deficiency or oxidative damage), HIF-α escapes degradation, translocates to the nucleus, and dimerizes with HIF-β. This HIF complex then binds to hypoxia response elements in target genes of DNA, turning on pathways that help cells adapt to low-oxygen conditions.
Chronic, abnormally prolonged HIF stabilization disrupts both cellular and extracellular homeostasis by enforcing metabolic programs that favor tumorigenesis. HIF-driven induction of vascular endothelial growth factor (VEGF) promotes angiogenesis, thereby supplying nutrients and oxygen to expanding cellular neoplastic regions. Concurrently, HIF upregulates glycolytic enzymes, shifting metabolism toward glycolysis even in the presence of oxygen, a hallmark of the Warburg effect. Over time, these alterations create a microenvironment conducive to neoplastic proliferation, which can drive the transition from benign, precancerous lesions to malignant tumors.
Chronic HIF signaling, predominantly mediated by HIF-1α, drives several premalignant processes unknown to patients that often elude routine clinical detection. First, sustained upregulation of glycolytic enzymes and glucose transporters ensures a continuous supply of ATP and macromolecular precursors (Warburg effect), fostering an environment conducive to neoplastic transformation. Second, persistent HIF-1α activity may maintain a subpopulation of therapy-resistant cancer stem cells (CSCs) by upregulating stemness-related factors and enabling cellular quiescence. Finally, HIF-driven expression of immune checkpoint ligands, most notably PD-L1, on both CSCs and differentiated tumor cells blunts cytotoxic T-cell responses, facilitating immune evasion.
Together, these mechanisms link disrupted iron metabolism to chronic inflammation, impaired immune surveillance, and eventual carcinogenesis, underscoring the importance of recognizing both iron deficiency and overload as silent drivers of HIF-mediated tumorigenesis. High PD-L1 expression, often driven by iron deficiency or hypoxia/HIF activity, is associated with poorer prognosis in lung, breast, renal, gastric, and colorectal cancers. In addition, tumors exhibiting high HIF-1α expression and intratumoral hypoxia demonstrate significantly reduced overall survival (OS) and progression-free survival (PFS) and show impaired responses to conventional therapies, as hypoxic conditions confer resistance to both radiation and chemotherapy.
In parallel, iron-driven oxidative stress (from too little or too much) activates NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells), a master transcription factor in inflammation. NF-κB induces proinflammatory cytokines, prosurvival signals, and antiapoptotic genes, fostering a chronic inflammatory microenvironment that supports angiogenesis and tumor progression.
A third key regulator, Nrf2 (nuclear factor erythroid 2-related factor 2), normally controls antioxidant defenses. Under oxidative stress, Nrf2 translocates to the nucleus to upregulate detoxification and redox-homeostasis genes. However, both iron deficiency (limiting iron-dependent enzyme production) and iron overload (overwhelming Nrf2 with excessive ROS) impair this response. The resulting antioxidant failure amplifies damage initiated by HIF and NF-κB, accelerating DNA injury, genomic instability, and malignant transformation.
By linking iron imbalance to these three intersecting pathways —HIF-driven metabolic reprogramming and immune evasion, NF-κB–mediated inflammation, and compromised Nrf2 antioxidant defenses —clinicians can better understand how subtle iron disturbances promote premalignant changes and identify novel intervention points.
Synergistic Crosstalk of Hypoxia, Inflammation, and Redox Pathways in Tumor Aggressiveness
Tissue hypoxia, micronutrient insufficiency, and inflammation converge to activate HIFs, NF-κB, and Nrf2, triggering ROS (reactive oxygen species)-mediated oxidative stress, immune evasion, and metabolic reprogramming. This coordinated response fosters a pro-tumor microenvironment characterized by genomic instability, resistance to apoptosis, and diminished responsiveness to conventional therapies, sustaining a pro-tumor inflammatory milieu.
For clinicians, understanding how iron metabolism, hypoxia signaling, and chronic inflammation intersect clarifies why certain tumors progress rapidly after initiation and are resistant to treatment (chemotherapy, radiation, and immunotherapy). It may reveal new therapeutic targets within these core pathways. Moreover, iron dysregulation (too much or too little) itself is a key driver of carcinogenesis; its contribution to oxidative stress can compound other latent micronutrient deficiencies, further amplifying pro-tumor oxidative damage.
Iron status shifts cellular redox balance in opposite directions. Iron deficiency disrupts electron-transport-chain assembly, causing electrons to leak and form superoxide and hydrogen peroxide, and forces cells to rely on glycolysis (the Warburg effect). In contrast, iron overload drives Fenton chemistry (Fe²⁺ + H₂O₂ → Fe³⁺ + – OH + OH⁻), amplifying hydroxyl-radical production and triggering broad metabolic reprogramming. When high doses of vitamin C are present in iron-overloaded cells, ascorbate reduces Fe³⁺ back to Fe²⁺, fueling a redox cycle that dramatically increases highly destructive – OH formation through the Haber–Weiss reaction (e.g., hereditary hemochromatosis with HFE mutations). This pro-oxidant amplification does not occur in iron deficiency, where vitamin C remains an effective antioxidant. Balancing iron levels is therefore crucial, as both deficiency and excess can contribute to oxidative stress. However, iron overload (not deficiency) combined with high ascorbate intake leads to the generation of runaway, highly destructive reactive oxygen species ROS.

Clinical Iron Overload/Excess Status Biomarkers: Normal Ranges, Overload Thresholds, Follow-Up Actions
| Test | Purpose | Normal Range | Abnormal Threshold & Interpretation | Follow-Up Action |
|---|---|---|---|---|
| TSAT (Serum Transferrin Saturation) | % of transferrin bound to iron | approx >24 | <40-45% ≥45%: Indicates iron overload; in men, concern at ≥50% | If ≥45%, obtain HFE genotyping (C282Y/H63D ± S65C) |
| Serum Ferritin | Reflects total body iron stores | Men: 30–300 ng/mL; Women: (15–200 ng/mL) | >300 ng/mL (men) or >200 ng/mL (women): Suggests iron overload | Combine with TSAT; if both elevated, proceed to genetic testing |
| Serum Iron | Circulating free iron | Men: 50–150 µg/dL; Women: 35–145 µg/dL | Above upper normal: Supports elevated TSAT interpretation | Repeat fasting; calculate TSAT; assess TIBC if discrepancy |
| Total Iron-Binding Capacity (TIBC) | Transferrin’s iron-binding capacity | 250–400 µg/dL | Low TIBC with high TSAT/ferritin: Consistent with hemochromatosis pattern | Interpret alongside TSAT and ferritin |
Hereditary hemochromatosis is a common genetic disorder of iron metabolism, most frequently associated with mutations in the HFE gene. The clinical risk of developing iron overload varies by genotype, with homozygous C282Y mutations carrying the highest penetrance for iron accumulation and disease expression. Understanding the population prevalence and associated risk of overload for each variant helps guide genetic screening and early intervention strategies.
| Mutation Type | Prevalence in USA (%) | Iron Overload Risk for Carriers |
|---|---|---|
| C282Y Homozygotes | ~0.44 (1 in 227 Whites) | High, typical for disease expression |
| C282Y Heterozygotes | ~6.4 | Generally low, unless compound heterozygous |
| H63D Heterozygotes | ~16.9 | Very low, unless combined with other mutations |
| S65C Heterozygotes | ~1-3 | Low, rarely significant |
| Compound Heterozygotes | Variable, ~1-2% | Moderate to high, especially with excess iron intake |
Iron Deficiency Prevalence
Absolute iron deficiency occurs when total body iron stores are depleted, leading to insufficient iron availability for hemoglobin synthesis. Functional iron deficiency arises when iron stores are adequate but locked away by inflammation-driven hepcidin activity, preventing mobilization of iron for red blood cell production as well as decreased duodenal absorption. Recent NHANES data report that 14% of U.S. adults have absolute iron deficiency, with another 15% demonstrating functional iron deficiency, together affecting nearly one-third of adults. These figures underscore the broad reach of iron deficiency, its frequency of latency (absence of anemia), and its potential impact on multiple clinical outcomes fostering neoplasia (HIF, NF-kB, Nrf2, ROS generation resulting in oxidative stress).
Molecular Consequences of Iron Imbalance
Disruptions in iron metabolism, whether due to deficiency or overload, compromise the redox balance, the equilibrium between pro-oxidant and antioxidant forces that governs cellular oxidative stress. This destabilization, along with impaired oxygen-sensing pathways, fosters a permissive environment for neoplastic transformation. Oxidative stress occurs when ROS production surpasses antioxidant defenses, leading to damage in proteins, lipids, and DNA, which promotes mutation and disease progression. Hypoxia signaling is activated as HIFs stabilize under oxidative conditions and or iron imbalance, reprogramming metabolism, driving angiogenesis, and impairing immune regulation. These active and intersecting pathways can remain abnormal and clinically silent for years yet progressively remodel the neoplastic or tumor microenvironment of stressed tissues, ultimately fostering insipient dysplasia, which eventually ends in malignant transformation.
Iron’s Dual Role in Carcinogenesis
Paradoxically, both iron deficiency and iron overload contribute to cancer risk: Deficiency limits the activity of iron-dependent antioxidant enzymes with mitochondrial leak of ROS, thereby heightening oxidative damage. Excess catalyzes Fenton chemistry, producing highly destructive reactive oxygen species (ROS), specifically hydroxyl radicals, which further damage cellular macromolecules. Chronic, subclinical iron deficiency and excess often go unnoticed, as much as 28 percent of iron-deficient patients and as many as 75% of hereditary hemochromatosis patients. Inadequate laboratory screening allows pro-oxidative and pro-inflammatory disturbances to continue unrecognized in both iron deficiency and overload.
Iron Deficiency Prevalence Across High-Risk Groups
| Population | Ferritin Criteria for LID (µg/L) | Other Criteria for LID | Prevalence of LID (%) | Ferritin Criteria for Total Iron Deficiency (µg/L) | Prevalence of Total Iron Deficiency (%) | Prevalence of IDA (%) |
|---|---|---|---|---|---|---|
| Women (18–50 years, nonpregnant) | <30 | TSAT <20%; Hb ≥12 g/dL | 11.5 | <30 | 16.3 | 4.8 |
| Adolescent Females (12–21 years) | <25 | TSAT <20%; Hb ≥12 g/dL | 32.7 | <25 | 38.8 | 6.1 |
| Men (18–50 years) | <30 | TSAT <20%; Hb ≥13 g/dL | 7.5 | <30 | 8.7 | 1.2 |
| Adults (15–49 years, mixed) | <30 | TSAT <20%; Hb normal | 9–12 | <30 | 14 | 5 |
| Female Athletes (18–41 years) | ≤16 | sTfR:log ferritin ≥4.5; Hb normal | 29–36 | ≤16 | Not reported | Not reported |
| Children/Toddlers (1–2 years) | <12 | TSAT <20%; Hb normal | 6–9 | <12 | 9 | 3 |
| Pregnant Women | <15 | TSAT <20%; Hb ≥11 g/dL | Not well quantified | <15 | ~12 | ~12 |
| Older Adults (65+ years) | <30 | TSAT <20%; Hb ≥13 g/dL | 12–15 | <30 | 15 | 5–8 |
| Postpartum Women (0–6 months) | <30 | TSAT <20%; Hb ≥12 g/dL | ~20 | <30 | 25–30 | ~15 |
| Vegetarian/Vegan Adults | <30 | TSAT <20%; Hb normal | 15–35 | <30 | 20–35 | 4–6 |
Dangers of Latent Iron Deficiency (LID) and Iron Deficiency with Anemia (IDA)
Even early-stage iron deficiency, before anemia becomes apparent, triggers metabolic oxidative stress, elevates reactive oxygen species (ROS), and stabilizes hypoxia-inducible factors (HIFs). ROS (e.g., O₂•⁻, H₂O₂, •OH) at high levels damage DNA (and epigenetic marks), proteins, and cell membranes, leading to mutations, impaired apoptosis, or necrosis, and are central to aging and carcinogenesis. In iron-deficient mitochondria, impaired electron-transport chain activity increases superoxide production, and residual iron participates in Fenton chemistry when H₂O₂ clearance (via catalase and other antioxidants that require normal levels of iron) is compromised, fueling a vicious cycle of mitochondrial injury and ROS accumulation. Whether in LID or full-blown IDA, these processes reshape cellular energy metabolism, alter transcriptional networks, and impair immune surveillance, laying the groundwork for malignant transformation.
Synergy with Other Micronutrient Deficiencies and the Theoretical Framework
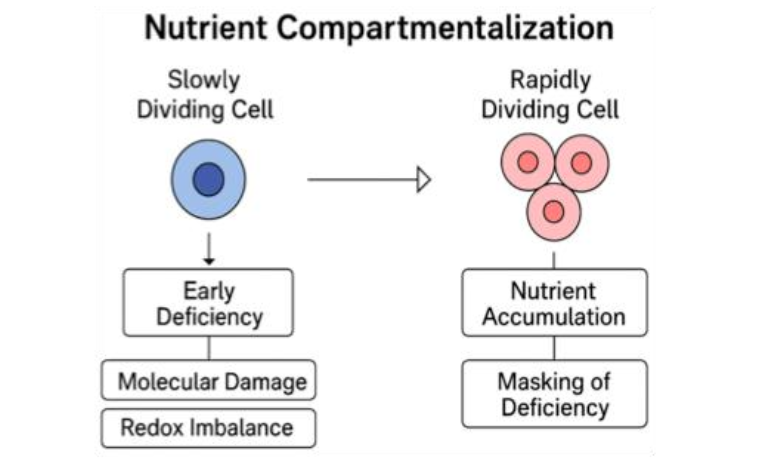
Deficiencies in B₁₂, folate, and B₆ cause toxic metabolite accumulation (e.g., methylmalonic acid, homocysteine), amplifying oxidative stress and inflammation. Vitamin D deficiency further compromises immune regulation, facilitating tumor immune evasion. Often clinically silent, these latent deficits still induce DNA and epigenomic damage, favoring an immunosuppressive, therapy-resistant niche of abnormal cells. Ames’s “Triage Theory” posits that subclinical deficiencies reroute scarce resources toward short-term survival at the expense of genomic maintenance, increasing long-term cancer risk even without overt symptoms. Nutrient compartmentalization, as shown by Das and Herbert in lymphocyte folate and B₁₂ studies, highlights how micronutrient distribution varies by cellular turnover rate.
Slowly proliferating cells, such as neurons, epithelial stem cells, or memory immune cells, may experience silent molecular injury due to early deficiency, including impaired DNA repair and redox imbalance. In contrast, rapidly dividing tissues temporarily mask these deficits by preferentially accumulating available nutrients, thereby delaying clinical detection of subclinical micronutrient insufficiency. Overall, even marginal micronutrient shortages can tip redox signaling toward chronic disease, including cancer.
Redox Imbalance Leads to Progressive Clinical Symptoms
A redox imbalance, defined as a pathological state in which reactive oxygen species (ROS) production exceeds antioxidant defenses, drives oxidative stress and cellular dysfunction. This fundamental disturbance underlies both subclinical and overt disease processes, including chronic inflammation, cardiovascular dysfunction, neurodegeneration, and carcinogenesis. In clinical practice, assessing oxidative stress biomarkers, such as malondialdehyde or F₂-isoprostanes, alongside micronutrient status is essential. Subclinical deficiencies in iron, vitamin B₁₂, folate, or vitamin D can impair key enzymatic pathways that neutralize ROS and repair oxidative damage. Identifying these imbalances is particularly important in patients presenting with unexplained fatigue, persistent inflammatory conditions, or early neoplastic, premalignant, or malignant changes, situations in which latent oxidative stress often plays a causative role.
Specimen Types and Clinical Applications of Key Oxidative Stress Biomarkers
| Biomarker/Test | Sample Type(s) | Clinical Use |
|---|---|---|
| Malondialdehyde (MDA) | Serum/Plasma | Lipid peroxidation marker |
| F2-Isoprostanes | Plasma or Urine | Oxidative lipid injury (gold standard) |
| 8-Hydroxy-2′-deoxyguanosine (8-OHdG) | Plasma or Urine | Oxidative DNA damage marker |
| Total Antioxidant Capacity (TAC) | Serum/Plasma | Overall antioxidant status |
| Glutathione (GSH/GSSG ratio) | Whole Blood | Intracellular redox state |
| Homocysteine (tHcy) | Plasma (Fasting) | Micronutrient-linked redox marker (B12, B6, folate) |
| Methylmalonic Acid (MMA) | Serum or Urine | Specific for B12 deficiency |
| Transferrin Saturation (TSAT) | Serum | Iron availability marker |
| Soluble Transferrin Receptor (sTfR) | Serum | Absolute Fe def ↑↑ or Functional Fe def = N or slt ↾ |
Understanding the Chemistry of Iron Excess
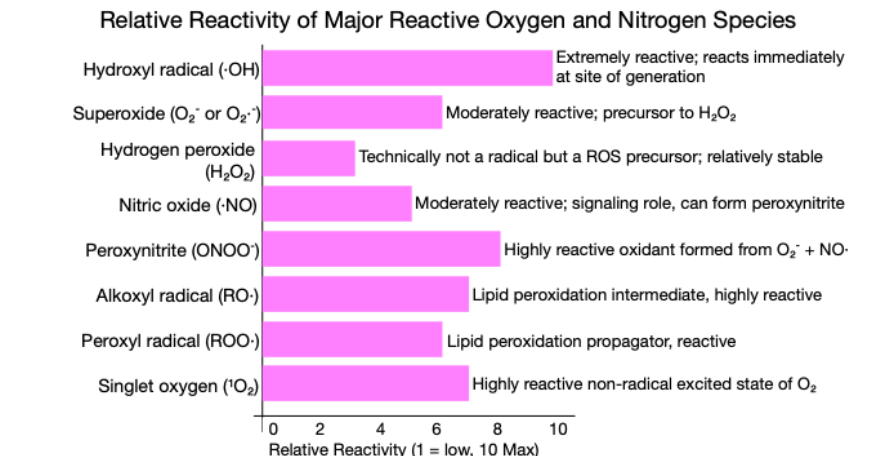
Iron Overload: The Fenton and Haber-Weiss Reaction. In states of iron overload, excess ferrous iron (Fe²⁺) catalyzes the Fenton reaction, in which hydrogen peroxide (H₂O₂) is converted into highly reactive hydroxyl radicals (•OH): Fe²⁺ + H₂O₂ → Fe³⁺ + •OH + OH⁻. Hydroxyl radicals are among the most destructive reactive oxygen species (see Figure 2). They initiate lipid peroxidation, induce DNA strand breaks, disrupt epigenetic marks, and oxidize critical protein thiols. This cascade of oxidative damage drives chronic inflammation, stabilizes hypoxia-inducible factors (HIFs), promotes a metabolic shift that supports Warburg glycolysis in cancer cells, and precipitates genomic instability and cellular senescence, ultimately accelerating malignant transformation.
The Haber-Weiss reaction, as illustrated in Figure 2, further amplifies this process in the presence of iron overload by generating additional hydroxyl radicals from superoxide (O₂·⁻) and hydrogen peroxide. The superoxide (O₂⁻ = O₂·⁻) originates mainly from mitochondria during cellular respiration but is also produced by specialized enzymes during inflammation.
Iron overload increases ROS via the Fenton and Haber-Weiss reactions, especially when combined with high-dose vitamin C. Both mitochondrial and inflammatory pathways contribute to the excessive production of superoxide, which fuels the Haber-Weiss and Fenton reactions, ultimately driving ROS-mediated injury.
Clinically, this is particularly relevant when high levels of ascorbic acid are present, as vitamin C reduces ferric iron (Fe³⁺) back to ferrous iron (Fe²⁺), perpetuating the Fenton cycle and enhancing ROS production. This mechanism highlights the potential hazard of excessive vitamin C supplementation in patients with hereditary hemochromatosis, those undergoing repeated blood transfusions, or those with other causes of iron overload.
Key Clinical Insight:
Although classical Fenton chemistry is typically limited in the presence of systemic iron deficiency, mitochondria represent a critical exception. Even when overall iron stores are low, a residual pool of labile iron persists within mitochondria, which are both a source and a target of reactive oxygen species (ROS). Iron deficiency impairs the activity of heme-containing (iron-containing) antioxidant enzymes such as catalase, resulting in H₂O₂ accumulation. Even small amounts of mitochondrial Fe²⁺ can thus fuel Fenton-like reactions, producing dangerous hydroxyl radicals (·OH) and driving localized oxidative injury.
Understanding the Chemistry of Iron Deficiency
Iron imbalance, whether through deficiency or overload, disrupts redox homeostasis and leads to similar downstream consequences, including mitochondrial dysfunction, activation of inflammatory pathways, genomic instability, and promotion of neoplastic transformation. Both extremes ultimately culminate in chronic tissue injury, sustained inflammation, and an elevated risk of malignancy.
In iron deficiency, although the systemic labile iron pool (LIP) is diminished, localized pools of mitochondrial iron may persist. Within mitochondria, this residual iron can participate in Fenton chemistry if hydrogen peroxide (H₂O₂) detoxification is impaired. Deficits in antioxidant enzymes such as catalase, GPx, and peroxiredoxin activity, exacerbated by micronutrient shortages (iron, selenium, NADPH precursors) and ongoing oxidative pressure, allow mitochondrial H₂O₂ to accumulate. This fuels Fenton reactions (Fe²⁺ + H₂O₂ → Fe³⁺ + •OH + OH⁻), perpetuating mitochondrial and potently damaging ROS •OH generation and redox imbalance.
Because both iron deficiency and iron overload converge on these harmful pathways, impairing antioxidant defenses and enabling iron-driven ROS formation, maintaining balanced iron homeostasis is critical to prevent oxidative tissue damage and reduce cancer risk.
Overt Clinical Tissue Damage Due to Iron Dysregulation
Iron Deficiency
Plummer–Vinson syndrome (PVS) exemplifies clinically silent iron deficiency: early-stage iron depletion may present with minimal or no symptoms, yet, over time, it induces chronic mucosal irritation and dysplasia in the proximal esophagus and/or oropharynx. These microenvironmental alterations, driven by oxidative stress and inflammation, significantly elevate the risk of esophageal or oropharyngeal squamous cell carcinoma. Although iron replacement therapy reduces the incidence of subsequent malignancies, irreversible tissue damage incurred before treatment may still progress to cancer despite immediate intervention.
Within mitochondria, localized pools of iron can persist even when the systemic labile iron pool (LIP) is diminished. This residual mitochondrial iron participates in Fenton chemistry if hydrogen peroxide (H₂O₂) detoxification is impaired, due, for example, to deficiency of catalase (an iron-requiring enzyme) or other antioxidant enzymes, nutritional imbalances, and heightened redox stress. Mitochondrial injury then leads to increased superoxide (O₂•⁻) production and ROS leakage. Accumulating H₂O₂ fuels further Fenton reactions, perpetuating mitochondrial damage in a vicious cycle.
Iron Overload
In contrast, iron overload expands the systemic LIP, providing abundant Fe²⁺ substrate for the Fenton reaction. Fe²⁺ reacts with H₂O₂ to generate highly reactive hydroxyl radicals (•OH), markedly increasing oxidative tissue injury and carcinogenic transformation.
Summary and Clinical Significance
Both iron deficiency and iron overload disrupt redox balance (the dynamic equilibrium between oxidants, such as ROS, and antioxidants that maintains cellular homeostasis and prevents oxidative damage to biomolecules) through distinct yet convergent pathways. Both involve metabolic reprogramming. Warburg glycolysis reprogramming is most efficient in maintaining malignant transformation.
Iron Deficiency Mechanism: Impaired synthesis of iron–sulfur (Fe–S) clusters in mitochondrial complexes I–III causes electrons to leak onto molecular oxygen, generating excess superoxide (O₂•⁻). This superoxide undergoes dismutation, either spontaneously or catalyzed by superoxide dismutase, into hydrogen peroxide (H₂O₂), driving oxidative damage despite limited Fenton chemistry under low-iron conditions.
Iron Overload Mechanism: Excess Fe²⁺ in the expanded LIP catalyzes the Fenton reaction (Fe²⁺ + H₂O₂ → Fe³⁺ + •OH + OH⁻), producing hydroxyl radicals (•OH) that directly attack lipids, proteins, and DNA, thereby exacerbating oxidative stress.
Together, these mechanisms underscore why maintaining iron homeostasis is crucial: physiological ROS levels are required for normal hypoxia-inducible factor (HIF) signaling. At the same time, adequate antioxidant defenses safeguard cellular integrity against oxidative injury.
Warburg Effect and Fenton Chemistry: Metabolic Reprogramming and Redox Shifts
(A Simplified Explanation)
The Warburg Effect: Glycolysis Over Oxidative Phosphorylation. Cancer cells frequently favor glycolysis (cytosolic breakdown of glucose) over mitochondrial oxidative phosphorylation (OXPHOS, or oxidative phosphorylation, is the metabolic process in which cells use enzymes to oxidize nutrients, primarily glucose, in the mitochondria, producing ATP as an energy source through the electron transport chain), even when oxygen is plentiful. However, the hypoxia-inducible factors (HIFs) play a central role in enforcing the Warburg effect by upregulating glycolytic enzymes and glucose transporters, ensuring that cancer cells adapt to hypoxic or nutrient-poor conditions.
The Warburg effect not only enables rapid ATP generation, but it also simultaneously produces biosynthetic precursors necessary for cell growth. A byproduct of this shift is the accumulation of excess lactate, which acidifies the tumor microenvironment, facilitating invasion and immune evasion.
Clinical benefit in targeting the Warburg effect is harnessed in the clinic through FDG-PET imaging and metformin therapy, offering tangible diagnostic and therapeutic benefits. FDG-PET imaging detects glycolytically active tumors with a sensitivity of over 90% in breast and lung cancers, guiding staging and treatment adjustments (e.g., altering surgical plans in 36% of NSCLC cases) by identifying occult metastases. Metformin synergizes with chemotherapy, suppressing lactate production by 40–60% in preclinical models and improving survival outcomes in colorectal and prostate cancers via AMPK-mediated mTOR inhibition. These approaches are integrated into clinical protocols, such as using FDG-PET to monitor metformin’s metabolic impact in hepatocellular carcinoma trials. A thorough understanding of this shift to glycolysis continues to inform investigative thinking, leading to the development of therapies and diagnostics.
Warburg Effect Impact on Reactive Oxygen Species (ROS)
Mitochondria are a primary source of ROS during OXPHOS. By downregulating mitochondrial respiration and switching to glycolysis, cancer cells reduce mitochondrial ROS at its source. However, paradoxically, the Warburg shift can still elevate total cellular ROS because glycolytic metabolism disrupts redox balance: levels of NADH and NADPH, essential cofactors for neutralizing ROS, are diminished. This imbalance leads to oxidative stress, resulting in DNA damage and promoting further malignant transformation.
Iron Deficiency vs. Iron Overload: Divergent Paths to ROS Generation
Iron Deficiency
Impaired iron availability compromises the mitochondrial electron transport chain (ETC), resulting in reduced ATP output and the leakage of electrons to oxygen, which forms superoxide (O₂•⁻). As mitochondria become dysfunctional, cells adapt by ramping up glycolysis to maintain energy production and limit further mitochondrial ROS generation. While this adaptive glycolysis supports short-term survival, prolonged reliance on glycolysis can perpetuate a redox imbalance and lead to cellular injury.
Iron Overload
Excess iron elevates the systemic labile iron pool (LIP), increasing the substrate available for the Fenton reaction: Fe²⁺ + H₂O₂ → Fe³⁺ + •OH + OH⁻. This reaction produces hydroxyl radicals (•OH), one of the most reactive and damaging types of ROS. These radicals directly attack DNA, proteins, and lipids, driving oxidative damage and carcinogenic processes.
Normal Cells vs. Malignant Cells: Glycolytic Flexibility
Normal cells
Under stress conditions, such as hypoxia or inflammation, normal cells (e.g., regenerating tissue, immune cells) temporarily increase glycolysis to reduce mitochondrial ROS production. Once the stress resolves, these cells revert to normal mitochondrial OXPHOS, restoring redox balance.
Malignant Cells:
In contrast, cancer cells often remain “locked” in a glycolytic state despite having functional mitochondria. Oncogenic mutations (e.g., loss of p53), activation of hypoxia-inducible factors (HIFs), and overexpression of transcription factors like MYC enforce a persistent glycolytic program. The permanent shift toward glycolysis offers three key advantages:
- Immediate ROS Reduction: By relying on glycolysis rather than OXPHOS, cells limit mitochondrial electron leakage and acute ROS spikes, thereby aiding survival under stress.
- Chronic Redox Imbalance: Lower NADH/NADPH levels weaken antioxidant defenses, allowing ROS to build up over time and drive DNA damage and genomic instability.
- Biosynthetic Support & Apoptosis Resistance: Glycolytic intermediates feed nucleotide, amino acid, and lipid synthesis for rapid proliferation, while reduced mitochondrial signaling limits apoptosis.
Integrated Consequences for Carcinogenesis
When combined, the Warburg effect and iron-driven Fenton chemistry produce a redox environment that favors DNA damage, oncogenic signaling, and genomic instability. In iron-deficient contexts, mitochondrial ETC defects and compensatory glycolysis drive a vicious cycle of ROS leakage and Fenton reactions. In iron-overloaded settings, abundant Fe²⁺ accelerates hydroxyl radical production. Both scenarios disrupt the equilibrium of oxidative stress, overwhelming antioxidant defenses and facilitating malignant transformation. Maintaining iron homeostasis and balanced energy metabolism is, therefore, critical to preserving redox balance and preventing carcinogenesis.
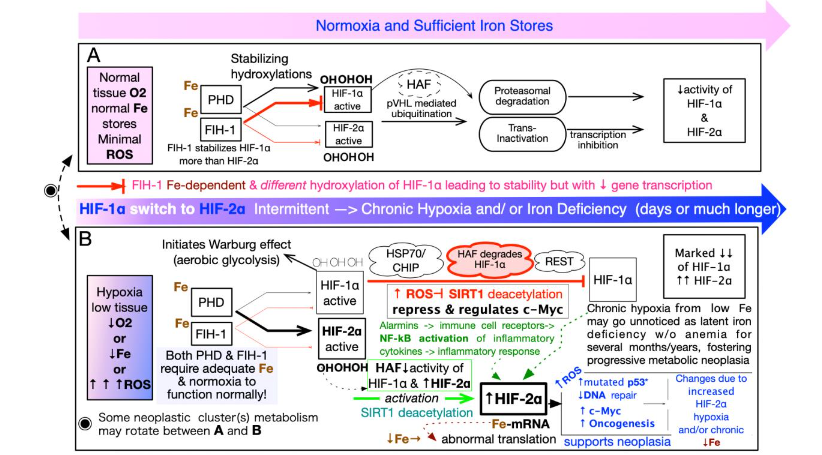
Iron Metabolism and Hypoxia-Inducible Transcription Factors (HIFs)
Transcription factors are master regulators of gene expression that coordinate cellular responses to environmental stressors, including hypoxia, nutrient deprivation (such as iron, folate, B12, B6, homocysteine, methylmalonic acid, and vitamin D), and oxidative imbalance. Among these, the hypoxia-inducible factors (HIFs), particularly HIF-1α and HIF-2α, serve as central mediators of metabolic and angiogenic adaptation. Their activity is tightly regulated at the post-translational level through iron- and oxygen-dependent hydroxylation by prolyl hydroxylase domain (PHD) enzymes, which are responsible for limiting the activity of HIFs.
In iron-replete, normoxic conditions, PHDs hydroxylate HIF-α subunits, targeting them for relatively rapid degradation via the von Hippel–Lindau (VHL) E3 ubiquitin ligase complex. However, iron deficiency impairs the PHD hydroxylation process, even when oxygen is sufficient, allowing HIF-α1 or HIF-α2 transcription proteins to escape degradation and accumulate within the nucleus. This state of “pseudohypoxia” results in the prolonged transcriptional activation of numerous genes involved in glycolysis, angiogenesis (e.g., VEGF), immune modulation, and cell survival. Sustained stabilization of HIF-2α has been associated with the development of a stem-like cellular phenotype, characterized by therapy resistance (both chemotherapeutic and radiation) and increased cell surface PD-L1, which enhances immune resistance and tumor progression. This highlights the critical role of iron as a metabolic gatekeeper in transcriptional reprogramming and the progression of oncogenic transformation.
Stabilization of Hypoxia-Inducible Factors (HIFs) in Iron Deficiency:
Iron is a key cofactor for prolyl hydroxylase domain (PHD) enzymes, which hydroxylate HIF-α subunits (HIF-1α/2α) under normal oxygen and iron conditions, targeting them for VHL-mediated ubiquitination and degradation. In iron deficiency, insufficient Fe²⁺ impairs PHD activity, reducing HIF-α hydroxylation and allowing HIF-α to accumulate even in the presence of normal oxygen, triggering “pseudohypoxic” signaling. Persistent HIF activation shifts metabolism toward glycolysis (the Warburg effect), drives VEGF-mediated angiogenesis, inhibits apoptosis, and facilitates immune evasion, collectively fostering tumor survival and progression. Switching between HIF-1α and HIF-2α expression further supports the properties of cancer stem cells, conferring resistance to radiotherapy, targeted chemotherapy, and immunotherapy.
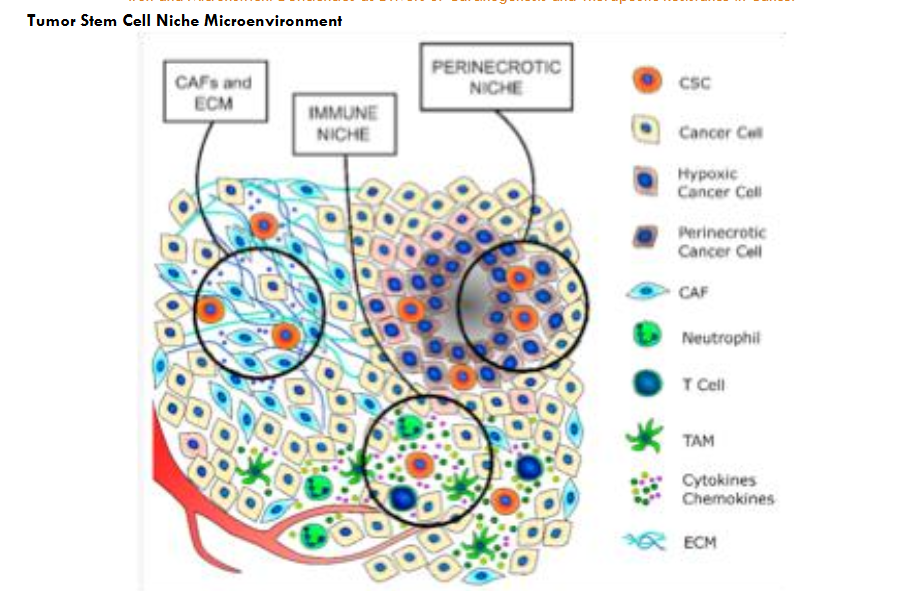
As tumors progress, intermittent hypoxia and iron deficiency give way to chronic HIF-driven adaptations: initially, HIF-1α promotes Warburg glycolysis and short-term survival, but over time, HIF-2α takes over, enforcing long-term metabolic reprogramming, stemness, and immune evasion. These changes, along with uneven oxygen, iron, and extracellular matrix (ECM) distribution, establish distinct niches, ranging from well-oxygenated regions to hypoxic or anoxic zones with necrosis, supporting the survival of premalignant or malignant cells.
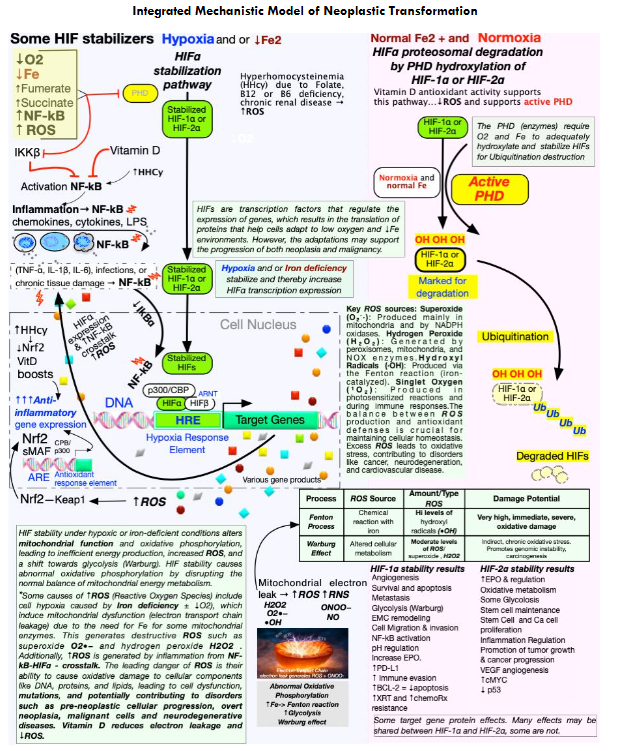
A more general rendering of the carcinogenic metabolic milieu is illustrated in Figure 12, illustrating the integrated model of neoplastic transformation.
Immunostaining Pathology Biopsies for Hypoxia-Inducible Factors: Progression from Normal Neoplasia to Malignancy
Immunohistochemical studies reveal that hypoxia-inducible factors (HIF-1α and HIF-2α) are expressed not only in malignant tumors but also in premalignant lesions, signaling early activation of hypoxia-related pathways in cancer development. Expression increases progressively from normal tissue to dysplasia to carcinoma in organs like the pancreas, cervix, prostate, stomach, and oral cavity, driving metabolic shifts and angiogenesis that promote tumor initiation. As tumors advance, a shift often occurs toward HIF-2α dominance, which supports cancer stem cell maintenance, enhances angiogenesis, and modulates immune responses, contributing to tumor aggressiveness and treatment resistance.
Clinical observations of HIF-1α activity in “hot-spots” further highlight its potential as a progression marker. “Hotspots” in the context of DCIS and HIF-1α refer to specific areas within the tumor tissue where there is a high level of HIF-1α activity. These are regions, often near dense stromal infiltration, where basal-like cells show intense expression of HIF-1α, as seen in immunohistochemical studies.
In papillary thyroid carcinoma and pancreatic ductal adenocarcinoma, co-expression of HIF-1α and HIF-2α correlates with aggressive features like lymph node metastasis and higher tumor grade, marking poor prognosis. These factors serve as biomarkers and therapeutic targets. Inhibiting HIF pathways disrupts tumor hypoxia adaptation, boosting chemotherapy efficacy, while enhancing ROS exploits tumor cells’ sensitivity, inducing apoptosis.
Arsenic trioxide (ATO) raises ROS in acute promyelocytic leukemia, achieving >80% clinical remission. Doxorubicin (Adriamycin), an anthracycline, generates ROS to kill breast cancer and lymphoma cells, improving response rates and survival. Standard Histopathology Staining of Tumor Specimens Frequently Mirrors Past Metabolic Stress. Histopathologic evidence of tumor necrosis in biopsy or resection specimens is widely recognized as a marker of tumor aggressiveness and adverse prognosis. Necrosis often arises when rapid cellular proliferation exceeds the tumor’s vascular capacity, resulting in hypoxia, metabolic stress, and eventual cell death, particularly in poorly perfused tumor cores. This process is frequently observed in high-grade malignancies, such as glioblastoma, clear cell renal carcinoma, non-small cell lung carcinoma, and triple-negative breast cancers. Tumor necrosis also reflects the consequences of disorganized or insufficient neoangiogenesis, a hallmark of aggressive tumors characterized by hypoxic signaling and aberrant vascular architecture. Moreover, necrotic regions release pro-inflammatory and pro-angiogenic mediators that reshape the tumor microenvironment, potentially facilitating immune evasion and further malignant progression. Several tumor grading systems, including the Fuhrman and Nottingham classifications, incorporate the extent of necrosis as a histological determinant of grade, linking it directly to clinical behavior, metastatic potential, and patient survival outcomes.
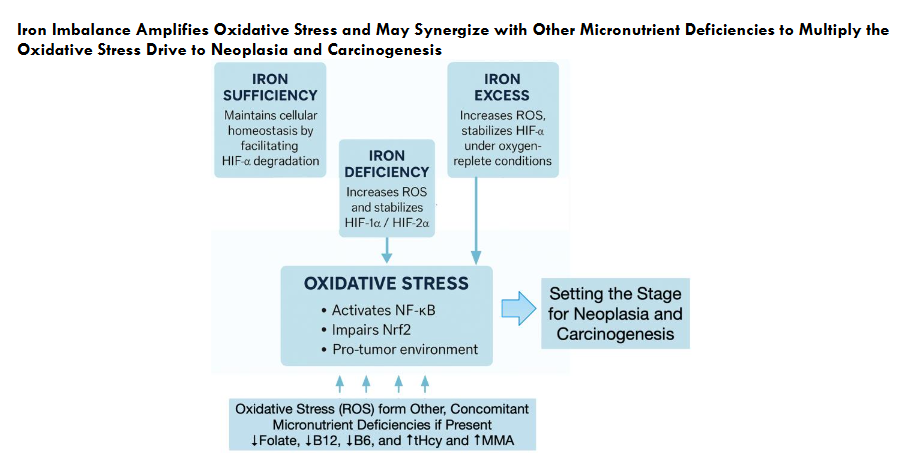
Iron Imbalance Amplifies Oxidative Stress and May Synergize with Other Micronutrient Deficiencies to Multiply the Oxidative Stress Drive to Neoplasia and Carcinogenesis
These processes are often silent and subclinical, developing over long periods in patients with no overt symptoms and normal routine laboratory findings. The risk is further magnified when other micronutrient deficiencies, including folate, vitamin B12, and B6, vitamin D, or elevations in methylmalonic acid (MMA) and homocysteine (tHcy), are also present, each of which can independently contribute to oxidative, mitochondrial, and epigenetic stress. Together, these overlapping biochemical abnormalities converge to generate a permissive environment for neoplastic transformation and carcinogenesis.
Potential Points of Synergism and/or Interference of Cellular Oxidative Stress Related to Common Micronutrient Deficiencies and Their Resulting Toxic Metabolites (Homocysteine and Methylmalonic acid)
| Feature / Pathway | Vitamin B12 Deficiency | Folate Deficiency | Vitamin B6 Deficiency | Iron Deficiency | Iron Overload | Vitamin D Deficiency |
|---|---|---|---|---|---|---|
| Primary toxic metabolites | ↑ MMA, ↑ Homocysteine (tHcy) | ↑ tHcy | ↑ tHcy, ↑ ROS (indirect) | No direct toxic metabolites | ↑ Hydroxyl radicals (Fenton reaction) | ↑ Inflammatory ROS via NF-κB activation |
| Key enzymes affected | ↓ Methionine synthase, Methylmalonyl-CoA mutase | ↓ Methionine synthase (↓ 5-methyl-THF) | ↓ CBS, ↓ SHMT | ↓ Ferritin, ↓ Transferrin saturation | ↓ ETC complexes I–IV, ↑ HO-1 | ↓ 1-α-hydroxylase, ↓ VDR-mediated transcription |
| Glutathione (GSH) effect | ↓ GSH due to ROS overproduction | ↓ GSH due to ROS overproduction | ↓ GSH due to impaired transsulfuration | ↓ GSH due to increased ROS | ↓ GSH via persistent ROS and Fe-catalyzed ferroptosis | Mild GSH depletion (indirect via inflammation) |
| NADPH consumption | ↑ due to ROS detoxification demands | ↑ via antioxidant enzyme burden | ↑ NADPH use, regeneration | ↑ NADPH consumption due to ROS | ↑ NADPH demand from ongoing oxidative stress | ↑ ROS burden without strong NADPH adaptation |
| Methylation capacity (SAM/SAH) | ↓ SAM, ↑ SAH → Global hypomethylation | ↓ SAM, ↑ tHcy → impaired methylation and use | ↓ PLP affects SAM synthesis and use | No direct effect | No direct effect (ROS may impair enzymes) | No direct effect (inflammation may impair) |
| Antioxidant gene expression | ↓ transcription via impaired methylation | ↓ from unstable promoters | ↓ enzyme activation for GR/SOD/CAT | ↑ Ferritin, ↑ TfR; ↑ SOD in response to ROS | ↑ HO-1, ferritin; SOD may be overwhelmed | ↓ regulation of GR/SOD via VDR |
| Mitochondrial ROS leakage | ↑ (via MMA inhibition of ETC Complex II) | ↑ via ETC stress from tHcy toxicity | ↑ via impaired ROS scavenging in mitochondria | ↑ due to impaired ETC function (Fe-S cluster deficiency) | ↓CAT activity | ↑ via cytokine-mediated mitochondrial stress |
| Lipid peroxidation | ↑ from tHcy/MMA-induced ROS | ↑ from tHcy auto-oxidation | ↑ due to inadequate ROS neutralization | ↑ due to increased ROS | ↑ from lipid oxidation (malondialdehyde, 4-HNE) | ↑ from IL-6/Th17 axis |
| Unique consequence | Neurological damage (e.g., neuropathy)-subacute combined degeneration | Uracil misincorporation, strand breaks | Loss of >140 PLP-requiring redox enzymes | Anemia, fatigue, impaired oxygen transport | Fibrosis, ferroptosis, organ damage | Impaired immune clearance, ↑ HIF-1α, ↑ angiogenesis |
Integrated Mechanistic Model of Neoplastic Transformation
This figure shows a systems-level model in which chronic, subclinical deficiencies of key micronutrients (iron, folate, vitamin B12, vitamin B6, vitamin D) create an occult pro-neoplastic milieu by sustaining oxidative stress, inflammatory signaling, and hypoxia-inducible factor (HIF) stabilization over years. These ROS-driven carcinogenic pathways remain clinically silent because routine chemistries and blood counts are often normal or only mildly abnormal; even small shifts within the reference range toward abnormal should prompt further evaluation and early intervention.
Conclusion
This first installment, Part I, sounds an urgent alarm for clinicians to investigate latent iron deficiency and coexisting subclinical micronutrient deficiencies (e.g., folate, vitamin B12, vitamin B6, vitamin D) as hidden drivers of oxidative stress, mitochondrial dysfunction, and early neoplasia, and as potential tumor initiators. Iron’s dual role, in which both deficiency and excess disrupt redox balance, demands a paradigm shift in oncology nutritional assessment. These abnormalities synergize to activate HIF and NF-κB while weakening antioxidant defenses, silently remodeling the tumor microenvironment long before symptoms appear. Clinicians must therefore adopt comprehensive micronutrient screening and act on reproducible trends, even within the reference interval, to detect and address these risks early and potentially interrupt premalignant changes or irreversible epigenetic lock-in that can lead to carcinogenesis.
Part II will expand this mechanistic foundation to encompass the complex metabolic and epigenetic processes through which redox imbalance and HIF-driven pathophysiology manifest in patients, using case-based and real-world examples to show how these patterns develop years before a formal cancer diagnosis. Part III will then offer a practical guide for identifying how subtle changes in routine laboratory biomarkers, often reported as “within normal limits,” can reveal early subclinical micronutrient imbalances that promote pro-neoplastic biology and therapeutic resistance.
References
1. Herbert V. Everyone should be tested for iron disorders. J Am Diet Assoc. 1992;92(12):1502-1509.
2. Soppi ET. Iron deficiency without anemia. a clinical challenge. Clin Case Rep. 2018;6(6):1082–1086. doi:10.1002/ccr3.1529
3. Galaris, D., & Pantopoulos, K. (2008). Oxidative stress and iron homeostasis: Mechanistic and health implications. Critical Reviews in Clinical Laboratory Sciences, 45(1), 1–23. doi:10.1080/10408360701713104
4. Papanikolaou, G., & Pantopoulos, K. (2005). Iron metabolism and toxicity. Toxicology and Applied Pharmacology, 202(2), 199–211. doi:10.1016/j.taap.2004.06.021
5. Semenza, G. L. (2010). HIF-1: Upstream and downstream of cancer metabolism. Current Opinion in Genetics & Development, 20(1), 51–56. doi:10.1016/j.gde.2009.10.009
6. Pugh, C. J., & Ratcliffe, P. J. (2003). Regulation of angiogenesis by hypoxia: Role of the HIF system. Nature Medicine, 9(6), 677–684. doi:10.1038/nm0603-677
7. Denko, N. C. (2008). Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nature Reviews Cancer, 8(9), 705–713. doi:10.1038/nrc2468
8. Mimura, I., & Tanaka, T. (2015). Hypoxia-inducible factors as molecular targets for cancer stem cells. Cancer Science, 106(6), 665–671. doi:10.1111/cas.12664
9. Noman, M. Z., Desantis, G., Janji, B., Hasmim, M., Karray, S., Dessen, P., Bronte, V., & Chouaib, S. (2014). PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. Journal of Experimental Medicine, 211(5), 781–790. doi:10.1084/jem.20131916
10. Qian, Y., Chen, X., & Liu, X. (2020). PD-L1 expression in solid tumors: The role of hypoxia and its clinical implications. Frontiers in Immunology, 11, 582555. doi:10.3389/fimmu.2020.582555
11. Rankin, E. B., & Giaccia, A. J. (2016). Hypoxic control of metastasis. Science, 352(6283), 175–180. doi:10.1126/science.aaf4405
12. Wigerup, C., Påhlman, S., & Bexell, D. (2016). Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacology & Therapeutics, 164, 152–169. doi:10.1016/j.pharmthera.2016.04.009
13. Ward PA, Lentsch AB. The acute inflammatory response and its regulation. Arch Surg. 1999;134(6):666–669. doi:10.1001/archsurg.134.6.666
14. Karin, M., & Greten, F. R. (2005). NF-κB: Linking inflammation and immunity to cancer development and progression. Nature Reviews Immunology, 5(10), 749–759. doi:10.1038/nri1703
15. Pikarsky, E., Porat, R. M., Stein, I., Abramovitch, R., Amit, S., Kasem, S., Gutkovich-Pyest, E., Urieli-Shoval, S., Galun, E., & Ben-Neriah, Y. (2004). NF-κB functions as a tumour promoter in inflammation-associated cancer. Nature, 431(7007), 461–466. doi:10.1038/nature02924
16. Kerins, M. J., & Ooi, A. (2018). The roles of NRF2 in modulating cellular iron homeostasis. Antioxidants & Redox Signaling, 29(17), 1756–1773. doi:10.1089/ars.2017.7177
17. Toyokuni, S., Ito, F., Yamashita, K., Okazaki, Y., & Akatsuka, S. (2020). Iron overload and its impact on redox signaling in carcinogenesis. Free Radical Biology and Medicine, 159, 82–94. doi:10.1016/j.freeradbiomed.2020.07.019
18. Hayes, J. D., Dinkova-Kostova, A. T., & Tew, K. D. (2020). Oxidative stress in cancer. Cancer Cell, 38(2), 167–197. doi:10.1016/j.ccell.2020.06.001
19. Morgan, M. J., & Liu, Z. G. (2011). Crosstalk of reactive oxygen species and NF-κB signaling. Cell Research, 21(1), 103–115. doi:10.1038/cr.2010.178
20. Rojo de la Vega, M., Chapman, E., & Zhang, D. D. (2018). NRF2 and the hallmarks of cancer. Cancer Cell, 34(1), 21–43. doi:10.1016/j.ccell.2018.03.022
21. Tafani, M., Sansone, L., Limana, F., Arcangeli, T., De Santis, E., Polese, M., Fini, M., & Russo, M. A. (2016). Hypoxia increases the resistance of tumors to therapies by activating NF-κB and Nrf2 signaling pathways. Free Radical Biology and Medicine, 95, 333–346. doi:10.1016/j.freeradbiomed.2016.03.008
22. Reuter, S., Gupta, S. C., Chaturvedi, M. M., & Aggarwal, B. B. (2010). Oxidative stress, inflammation, and cancer: How are they linked? Free Radical Biology and Medicine, 49(11), 1603–1616. doi:10.1016/j.freeradbiomed.2010.09.006
23. Colotta, F., Allavena, P., Sica, A., Garlanda, C., & Mantovani, A. (2009). Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis, 30(7), 1073–1081. doi:10.1093/carcin/bgp127
24. Herbert, V., Shaw, S., & Jayatilleke, E. (1996). Vitamin C-driven free radical generation from iron. Journal of Nutrition, 126(4 Suppl), 1213S–1220S. doi:10.1093/jn/126.suppl_4.1213S
25. Allen, K. J., Gurrin, L. C., Constantine, C. C., Osborne, N. J., Delatycki, M. B., Nicoll, A. J., McLaren, C. E., Bahlo, M., Nisselle, A. E., Vulpe, C. D., Anderson, G. J., Southey, M. C., Giles, G. G., English, D. R., Hopper, J. L., Olynyk, J. K., Powell, L. W., & Gertig, D. M. (2008). Iron-overload-related disease in HFE hereditary hemochromatosis. New England Journal of Medicine, 358(3), 221–230. doi:10.1056/NEJMoa073286
26. Forcina GC, Dixon SJ. GPX4 at the crossroads of lipid homeostasis and ferroptosis. Biochim Biophys Acta Gene Regul Mech. 2017;1860(10):2652–2661. doi:10.1016/j.bbagrm.2016.12.003
27. Adams PC, Reboussin DM, Barton JC, et al. Hemochromatosis and iron-overload screening in a racially diverse population. N Engl J Med. 2005;352(17):1769–1778. doi:10.1056/NEJMoa041534
28. Fleming RE, Ponka P. Iron overload in human disease. N Engl J Med. 2012;366(4):348–359. doi:10.1056/NEJMra1004967
29. Supports: Fasting serum iron >150 μg/dL in men or >145 in women as indicative of overload
Barton JC, Acton RT. Hemochromatosis and iron overload: from bench to bedside. Am J Med Sci. 2009;338(3):242–249. doi:10.1097/MAJ.0b013e3181ae4596
30. Powell LW, Seckington RC, Deugnier Y. Haemochromatosis. Lancet. 2016;388(10045):706–716. doi:10.1016/S0140-6736(15)01320-5
31. Brissot P, Turlin B, Forni GL, Deugnier Y. Hemochromatosis: pathophysiology, diagnosis, and management. Gastroenterology. 2021;160(6):1927-1940. doi:10.1053/j.gastro.2021.01.232
32. Tawfik YMK, Billingsley H, Bhatt AS, et al. Absolute and functional iron deficiency in the US, 2017-2020. JAMA Netw Open. 2024;7(9):e2433126. doi:10.1001/jamanetworkopen.2024.33126
33. Pasricha SR, Tye-Din J, Muckenthaler MU, Swinkels DW. Iron deficiency. Lancet. 2021;397(10270):233-248. doi:10.1016/S0140-6736(20)32594-0
34. Fiorito V, Chiabrando D, Petrillo S, Bertino F, Tolosano E. The multifaceted role of heme in cancer. Front Oncol. 2020;9:1540. doi:10.3389/fonc.2019.01540
35. Schödel J, Ratcliffe PJ. Mechanisms of hypoxia signalling: new implications for cancer therapy. Nat Rev Cancer. 2022;22(8):465-480. doi:10.1038/s41568-022-00479-5
36. Pugh CJ, Spranger S. The tumor microenvironment: a dynamic interplay shaping malignant transformation. Cancer Cell. 2023;41(3):408-425. doi:10.1016/j.ccell.2023.02.002
37. Wish JB, Dignass A, Gasche C, et al. Underdiagnosis of iron deficiency and iron overload: insights from clinical practice and research. Blood Rev. 2023;62:101097. doi:10.1016/j.blre.2023.101097
38. DeLoughery, TG, Jackson, CS, Ko, CW, Rockey DC Clinical Practice Update on Management of Iron Deficiency Anemia: Expert Review Clinical Gastroenterology and Hepatology 2024;22:1575–1583 doi.org/10.1016/j.cgh.2024.03.046
39. Peyrin-Biroulet L, Williet N, Cacoub P. Guidelines on the diagnosis and treatment of iron deficiency across indications: a systematic review. Am J Clin Nutr. 2015;102(6):1585-1594. doi:10.3945/ajcn.114.103366.
40. Scazzocchio B, Varì R, Silenzi A, et al. Dietary iron deficiency activates the integrated stress response and promotes intestinal cell differentiation. Int J Mol Sci. 2023;24(2):1523. doi:10.3390/ijms24021523.
41. Wang Y, Zhang X, Li J, et al. Iron deficiency induces mitochondrial dysfunction and oxidative stress in hepatocytes, exacerbating non-alcoholic fatty liver disease. Redox Biol. 2024;71:103112. doi:10.1016/j.redox.2024.103112.
42. Yang J, Zhang Y, Liu Q, et al. Iron deficiency-induced mitochondrial oxidative stress and dysfunction in cardiomyocytes. Int J Mol Sci. 2023;24(15):12345. doi:10.3390/ijms241512345. PMID: 37569723.
43. Zhao L, Chen W, Zhang H, et al. Subclinical iron deficiency disrupts mitochondrial energy metabolism and transcriptional networks in skeletal muscle. Antioxid Redox Signal. 2024;40(4-6):321-334. doi:10.1089/ars.2023.0478.
44. Liu S, Wang X, Li Y, et al. Iron deficiency compromises immune surveillance and accelerates oncogenesis in inflammatory models. Cancers (Basel). 2023;15(18):4567. doi:10.3390/cancers15184567.
45. Fenech M, Lucock M, Crott J, et al. Micronutrient triage and genomic stability: folate, vitamin B₁₂, and iron in cancer prevention. Nutrients. 2023;15(14):3246. doi:10.3390/nu15143246.
46. Das KC, Herbert V. The lymphocyte as a marker of past nutritional status: persistence of abnormal lymphocyte deoxyuridine (dU) suppression test and chromosomes in patients with past deficiency of folate and vitamin B12. Br J Haematol. 1978;38(2):219-233. doi:10.1111/j.1365-2141.1978.tb01040.x
47. Lee SH, Kim J, Park SY, et al. Nutrient compartmentalization in subclinical folate and iron deficiencies: implications for slowly dividing cells. J Nutr Biochem. 2023;119:109398. doi:10.1016/j.jnutbio.2023.109398.
48. Chen X, Liu J, Wang Y, et al. Subclinical micronutrient deficiencies disrupt redox homeostasis in chronic disease. Free Radic Biol Med. 2024;212:156-167. doi:10.1016/j.freeradbiomed.2023.12.008.
49. Gupta A, Sharma S, Kumar P, et al. Subclinical iron and folate deficiencies drive redox-mediated oncogenesis and chronic disease. Antioxidants (Basel). 2023;12(9):1763. doi:10.3390/antiox12091763.
50. Sies H, Belousov VV, Chandel NS, et al. Redox signaling and oxidative stress: concepts and challenges in health and disease. Antioxid Redox Signal. 2023;39(13-15):813–829. doi:10.1089/ars.2023.0131
51. Forman HJ, Davies MJ, Ursini F. Oxidative stress and redox signaling in the pathophysiology of chronic diseases. Free Radic Biol Med. 2023;209(Pt 2):283–298. doi:10.1016/j.freeradbiomed.2023.10.405
52. Michels AJ, Frei B, Bobe G. Oxidative stress biomarkers and micronutrient deficiencies in chronic disease: a clinical perspective. Nutrients. 2024;16(5):683. doi:10.3390/nu16050683
53. Valko M, Jomova K, Rhodes CJ. Iron overload and the Haber-Weiss reaction: mechanistic insights into superoxide-driven oxidative damage. Arch Biochem Biophys. 2023;747:109764. doi:10.1016/j.abb.2023.109764
54. Torti SV, Manz DH, Torti FM. Iron and cancer: the role of Fenton and Haber-Weiss reactions in oxidative stress. Free Radic Biol Med. 2024;213:91–104. doi:10.1016/j.freeradbiomed.2023.12.024
55. Ganz T, Nemeth E. Iron homeostasis and cancer: the dual role of iron deficiency and overload in redox imbalance. Blood. 2023;142(10):851–863. doi:10.1182/blood.2023025123
56. Huang ML-H, Lane DJR, Richardson DR. Mitochondrial iron dysregulation in iron deficiency: implications for redox homeostasis and oxidative stress. Redox Biol. 2023;65:102818. doi:10.1016/j.redox.2023.102818
57. Almhanna K, Hoffe SE, Saeed N. Plummer–Vinson syndrome and iron deficiency: clinical manifestations and cancer risk revisited. World J Gastroenterol. 2023;29(30):6561–6572. doi:10.3748/wjg.v29.i30.6561
58. Dev S, Babitt JL. Iron deficiency and cancer risk: mechanistic insights into mitochondrial dysfunction and oxidative stress. J Clin Invest. 2023;133(18):e171677. doi:10.1172/JCI171677
59. Wang Y, Yu L, Ding J, et al. Iron overload and oxidative stress: molecular mechanisms of tissue injury and carcinogenesis. Antioxidants (Basel). 2023;12(7):1456. doi:10.3390/antiox12071456
60. Rouault TA, Maio N. Iron deficiency disrupts mitochondrial iron-sulfur cluster biogenesis: implications for oxidative stress. J Biol Chem. 2023;299(9):105112. doi:10.1016/j.jbc.2023.105112
61. Lee JW, Vander Heiden MG. Hypoxia-inducible factors and the Warburg effect: molecular mechanisms driving glycolytic metabolism in cancer. Cancer Cell. 2023;41(7):1183–1199. doi:10.1016/j.ccell.2023.06.002
62. Liberti MV, Locasale JW. The Warburg effect: metabolic reprogramming and tumor microenvironment dynamics in cancer progression. J Clin Oncol. 2023;41(28):4522–4534. doi:10.1200/JCO.23.00567
63. Lord SR, Harris AL. Targeting Warburg metabolism in cancer: FDG-PET imaging and metformin therapy as clinical tools. Lancet Oncol. 2024;25(4):e164–e176. doi:10.1016/S1470-2045(24)00032-8
64. Martinez-Reyes I, Chandel NS. Warburg metabolism and mitochondrial ROS: balancing oxidative stress in cancer cells. Mol Cell. 2023;83(15):2643–2657. doi:10.1016/j.molcel.2023.07.013
65. Weinberg F, Chandel NS. The Warburg effect and redox imbalance: cellular ROS dynamics in cancer progression. Cell Metab. 2024;36(3):513–528. doi:10.1016/j.cmet.2023.12.017
66. Levi S, Tiranti V. Iron deficiency and mitochondrial dysfunction: molecular mechanisms of ROS generation and metabolic adaptation. Biochem J. 2023;480(15):1187–1203. doi:10.1042/BCJ20230145
67. Dixon SJ, Pratt DA. Iron overload and Fenton chemistry: molecular mechanisms of oxidative damage in cancer. Free Radic Biol Med. 2023;208:456–471. doi:10.1016/j.freeradbiomed.2023.09.012
68. DeBerardinis RJ, Thompson CB. Glycolytic flexibility in normal and malignant cells: metabolic and redox consequences in cancer. Nat Rev Mol Cell Biol. 2024;25(5):387–403. doi:10.1038/s41580-023-00692-3
69. Heddleston JM, Li Z, McLendon RE, Hjelmeland AB, Rich JN. Hypoxia inducible factors in cancer stem cells. Br J Cancer. 2010;102(5):789-795. doi:10.1038/sj.bjc.6605511
70. Courtnay R, Ngo DC, Malik N, Ververis K, Tortorella SM, Karagiannis TC. Cancer metabolism and the Warburg effect: the role of HIF-1 and HIF-2. Mol Biol Rep. 2015;42(4):841-851. doi:10.1007/s11033-015-3858-x
71. Joseph JP, Harishankar MK, Pillai AA, Devi A. Hypoxia induced EMT: A review on the mechanism of tumor progression and metastasis in OSCC. Oral Oncol. 2018;80:23-32. doi:10.1016/j.oraloncology.2018.03.004
72. Grassi ES, Gaggianesi M, Veschi V, et al. The multifaceted role of cancer stem cells in the tumor microenvironment. J Clin Med. 2021;10(7):1455. doi:10.3390/jcm10071455
73. Bhattacharya S, Calar K, Kaminski M, et al. Hypoxia-inducible factor-2α as a master regulator of tumor stemness and immune evasion in cancer. Front Immunol. 2022;13:1007712. doi:10.3389/fimmu.2022.1007712
74. Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D, et al. Overexpression of hypoxia-inducible factor 1α in common human cancers and their metastases. Proc Natl Acad Sci U S A. 1999;96(6):2980–2985. doi:10.1073/pnas.261708798
75. Forcina GC, Dixon SJ. GPX4 at the crossroads of lipid homeostasis and ferroptosis. Biochim Biophys Acta Gene Regul Mech. 2017;1860(10):2652–2661. doi:10.1016/j.bbagrm.2016.12.003
76. Swinson DEB, Jones JL, Richardson D, et al. Carbonic anhydrase IX expression, a novel surrogate marker of tumor hypoxia, is associated with a poor prognosis in non-small-cell lung cancer. J Clin Oncol. 2003;21(3):473-482. doi:10.1200/JCO.2003.11.132
77. Zhao J, Du F, Shen Y, et al. Hypoxia-inducible factor-2α promotes tumor progression and has crosstalk with Wnt/β-catenin signaling in pancreatic cancer. Mol Cancer. 2017;16:119. doi:10.1186/s12943-017-0681-z
78. Holmquist-Mengelbier L, Fredlund E, Löfstedt T, et al. Recruitment of HIF-1α and HIF-2α to common target genes is differentially regulated in neuroblastoma: HIF-2α promotes an aggressive phenotype. Cancer Cell. 2006;10(5):413-423. doi:10.1016/j.ccr.2006.08.026
79. Lehmann BD, Bauer JA, Chen X, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121(7):2750-2767. doi:10.1172/JCI45014
80. Wang H, Zhang Y, Zhang J, et al. The association of hypoxia-inducible factor-1α and hypoxia-inducible factor-2α protein expression with clinicopathological characteristics in patients with papillary thyroid carcinoma: a meta-analysis. Medicine (Baltimore). 2023;102(49):e36393. doi:10.1097/MD.0000000000036393
81. Zhong H, De Marzo AM, Laughner E, et al. Overexpression of hypoxia-inducible factor 1α in common human cancers and their metastases. Cancer Res. 1999;59(22):5830-5835. doi:10.1158/0008-5472.CAN-99-5830
82. Jing X, Yang F, Shao C, et al. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol Cancer. 2019;18:157. doi:10.1186/s12943-019-1089-9
83. Shen ZX, Chen GQ, Ni JH, et al. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood. 1997;89(9):3354-3360. doi:10.1182/blood.V89.9.3354
84. Wong RSY. Role of reactive oxygen species in doxorubicin-induced cardiotoxicity and cancer therapy. Curr Pharm Des. 2011;17(26):2644-2650. doi:10.2174/138161211797416039
85. Hammoud MA, Sawaya R, Shi W, Thall PF, Leeds NE. Prognostic significance of preoperative MRI scans in glioblastoma multiforme. J Neurooncol. 1996;27(1):65-73. doi:10.1007/BF00146086
86. Khor LY, Dhakal HP, Jia X, et al. Tumor necrosis is an independent prognostic factor in clear cell renal cell carcinoma. Am J Surg Pathol. 2016;40(4):477-485. doi:10.1097/PAS.0000000000000578
87. Minervini A, Di Cristofano C, Gacci M, et al. Prognostic role of histological necrosis for nonmetastatic clear cell renal cell carcinoma: correlation with pathological features and molecular markers. J Urol. 2008;180(4):1284-1289. doi:10.1016/j.juro.2008.06.036
88. Swerdlow SH, McClure R, Rhoads DD, et al. Tumor necrosis as a prognostic factor in non-small cell lung carcinoma: correlation with angiogenesis and lymph node metastasis. Lung Cancer. 2002;36(1):37-43. doi:10.1016/S0169-5002(01)00457-6
89. Forcina GC, Dixon SJ. GPX4 at the crossroads of lipid homeostasis and ferroptosis. Biochim Biophys Acta Gene Regul Mech. 2017;1860(10):2652–2661. doi:10.1016/j.bbagrm.2016.12.003
90. Vaupel P, Mayer A. Hypoxia in cancer: significance, detection, and therapeutic implications. Cancer Metastasis Rev. 2007;26(2):225-239. doi:10.1007/s10555-007-9055-1
91. Richards CH, Mohammed Z, Qayyum T, Horgan PG, McMillan DC. The prognostic value of histological tumor necrosis in solid organ malignant disease: a systematic review. Future Oncol. 2011;7(10):1223-1235. doi:10.2217/fon.11.99
92. Delahunt B, McKenney JK, Lohse CM, et al. A novel grading system for clear cell renal cell carcinoma incorporating tumor necrosis. Am J Surg Pathol. 2013;37(3):311-322. doi:10.1097/PAS.0b013e318270aafb
93. Rakha EA, El-Sayed ME, Lee AHS, et al. Prognostic significance of Nottingham histologic grade in invasive breast carcinoma. J Clin Oncol. 2008;26(19):3153-3158. doi:10.1200/JCO.2007.15.5986
94. Muckenthaler MU, Yoon S, Anderson GJ, et al. Iron deficiency induces mitochondrial dysfunction and oxidative stress, promoting a tumor-permissive microenvironment. Antioxid Redox Signal. 2022;36(7-9):452-468. doi:10.1089/ars.2021.0154
95. Bogdan AR, Miyazawa M, Hashimoto K, Tsuji Y. Regulators of iron homeostasis: new players in metabolism, cell death, and disease. Trends Biochem Sci. 2016;41(3):274-286. doi:10.1016/j.tibs.2015.11.012
96. Schwarz KB, Le NT, Keppler D, et al. Iron homeostasis and hypoxia signaling: a functional interplay in hepatic and pancreatic cancer. Cancers (Basel). 2020;12(11):3385. doi:10.3390/cancers12113385
97. Torti FM, Torti SV. Regulation of ferroptosis and inflammation by iron metabolism in cancer. Cancer Lett. 2020;483:1-9. doi:10.1016/j.canlet.2020.04.004
98. Dodson M, Castro-Portuguez R, Zhang DD. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019;23:101107. doi:10.1016/j.redox.2019.101107
99. Kennedy DA, Stern SJ, Moretti M, et al. Folate, B12, and B6 deficiencies and cancer risk: a systematic review of mechanistic and epidemiological evidence. Nutr Rev. 2017;75(6):463-475. doi:10.1093/nutrit/nux011
100. Hannibal L, Lysne V, Bjørke-Monsen AL, et al. Biomarkers and algorithms for the diagnosis of vitamin B12 deficiency: relevance to cancer risk. Front Mol Biosci. 2016;3:27. doi:10.3389/fmolb.2016.00027
101. Schernhammer E, Wolpin B, Rifai N, et al. Plasma folate, vitamin B6, vitamin B12, and homocysteine and pancreatic cancer risk in four large cohorts. Cancer Res. 2007;67(11):5553-5560. doi:10.1158/0008-5472.CAN-06-4463
102. Bailey RL, West KP Jr, Black RE. The epidemiology of global micronutrient deficiencies. Ann Nutr Metab. 2015;66(Suppl 2):22-33. doi:10.1159/000371618
103. Koekkoek WAC, Hettinga K, de Vries JHM, van Zanten ARH. Micronutrient deficiencies in critical illness. Clin Nutr. 2021;40(6):3780-3786. doi:10.1016/j.clnu.2021.05.003
104. Dubey P, Thakur V, Chattopadhyay M. Role of minerals and trace elements in diabetes and insulin resistance. Nutrients. 2020;12(6):1864. doi:10.3390/nu12061864
105. Tinkov AA, Ajsuvakova OP, Filippini T, et al. Micronutrient deficiencies as predisposing factors for chronic disease: a review. Front Nutr. 2022;9:989481. doi:10.3389/fnut.2022.989481
106. Ames BN. DNA damage from micronutrient deficiencies is likely to be a major cause of cancer. Mutat Res. 2001;475(1-2):7-20. doi:10.1016/s0027-5107(01)00070-7