






Thermodynamic Insights into Protein Dynamics in Medicine
Thermodynamic Insights into Protein Dynamics and Drug Development
Jawad Alzeer1, PhD
- College of Medicine and Health Sciences, Palestine Polytechnic University, Hebron, Palestine. Swiss Scientific Society for Developing Countries, Zürich, Switzerland.
Email: [email protected]
OPEN ACCESS
PUBLISHED: 31 August 2025
CITATION: Alzeer, J., 2025. Thermodynamic Insights into Protein Dynamics and Drug Development. Medical Research Archives, [online] 13(8).
https://doi.org/10.18103/mra.v13i8.6867
COPYRIGHT: © 2025 European Society of Medicine. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
DOI https://doi.org/10.18103/mra.v13i8.6867
ISSN 2375-1924
Abstract
Protein folding and misfolding play central roles in both health and disease, yet traditional structural analyses often fall short in explaining their functional consequences. This manuscript introduces a thermodynamic framework that integrates three molecular dimensions—chemical composition (constitution), stereochemistry (configuration), and conformational flexibility (conformation)—to better understand how proteins maintain function or drift toward dysfunction. By reframing potential energy and entropy as cooperative forces that govern structural transitions, this model provides insight into the dynamic behavior of proteins in physiological and pathological contexts. Correct folding involves a regulated increase in structural order, a reduction in conformational entropy, and a rise in internal potential energy that supports molecular precision. In contrast, disruptions in this balance can lead to misfolded proteins, aggregation, and disease states such as neurodegeneration, cancer, or immune dysfunction. The framework also highlights how flexible, disordered regions within proteins—often overlooked—can be targeted to design more selective, adaptive, and less toxic therapeutics. By linking molecular structure to biological outcome, this perspective offers a clinically relevant lens for advancing drug development, understanding disease mechanisms, and designing precision medicine strategies.
Keywords
Potential energy, Entropy, Protein conformation, Constitution–Configuration–Conformation, Intrinsically disordered proteins (IDPs), Drug design, Molecular flexibility.
Introduction
Biological systems, especially proteins, behave in ways that cannot be fully understood by looking at structure or energy in isolation. Proteins are dynamic molecules that shift between many shapes and states as they carry out their functions. These changes are not random—they are governed by the rules of thermodynamics, particularly the balance between potential energy and entropy. While entropy drives systems toward disorder and flexibility, potential energy stores the capacity for order, structure, and controlled interactions. These two forces shape how proteins fold, move, interact, and evolve.1-2 This interplay reflects a deeper principle; the Second Law of Thermodynamics, which states that in any natural process, total entropy tends to increase. In the context of proteins, this means that flexibility, motion, and disordered states are thermodynamically favored. However, life depends on maintaining structure and specificity. To slow the accumulation of entropy and preserve order, proteins use potential energy embedded in their chemical makeup. This energy allows proteins to resist complete disorder, enabling them to hold stable shapes while still remaining adaptable. Potential energy, then, can be seen as the thermodynamic force that opposes entropy—not by preventing it entirely, but by regulating it in ways that support biological function.
Proteins do not exist in a single static shape. They switch between folded, unfolded, misfolded, disordered, or ligand-bound forms—each with a different balance of energy and entropy.3-4 To understand this behavior, we introduce a structural model based on constitution, configuration, and conformation (CCC).5 Together, these three aspects offer a complete view of how proteins store energy, adapt their shapes, and manage thermodynamic forces.6
In this CCC framework, potential energy includes more than just bond energies or internal strain. It represents a protein’s stored ability to form specific interactions, resist deformation, or undergo controlled changes during binding or catalysis. This stored energy is essential for directing biological responses and supporting structured transitions. At the same time, entropy is not simply randomness. It reflects a protein’s freedom to explore different conformations, interact with its environment, and remain flexible in regions that require rapid adaptation. Some parts of a protein, such as loops or disordered regions, retain high entropy to allow movement and responsiveness, while others lock into low-entropy states to provide stability.7
This balance between entropy and potential energy is critical for health and disease. Too much rigidity can prevent proteins from adjusting to new signals, while too much flexibility may lead to loss of function or aggregation. In drug design, this balance becomes especially important. Drugs that over-stabilize proteins may block natural motions and cause resistance or toxicity.8-10 In contrast, ligands that interact with flexible, allosteric sites can fine-tune function without eliminating necessary dynamics. This is particularly relevant for targeting intrinsically disordered proteins (IDPs), which rely on entropy-rich states to carry out diverse and adaptable roles in the cell.11-12
By exploring these ideas, this manuscript offers a thermodynamic view of protein behavior grounded in the dual role of potential energy and entropy. Rather than viewing folding and function as purely structural events, we propose that they emerge from a regulated exchange between stored energy and flexible freedom. This perspective not only clarifies how proteins work, but also provides a helpful framework for drug discovery, disease understanding, and education—encouraging scientists to think thermodynamically, not just structurally, about life at the molecular level.
The Constitution, Configuration, and Conformation Framework
In living systems, potential energy is not just a theoretical concept—it is structurally encoded within the molecules themselves. Proteins, in particular, carry this energy not only in their chemical bonds but across multiple structural layers that shape their thermodynamic behavior. To understand how this energy is stored, distributed, and used, we introduce the Constitution, Configuration, and Conformation (CCC) framework—a structural model that captures how molecular identity and flexibility influence folding, function, and interaction.13-14
Constitution refers to the fundamental chemical makeup of a molecule—the types of atoms it contains and how they are connected. In proteins, this is the amino acid sequence. Certain residues, like cysteine, offer unique energy potential due to their ability to form disulfide bonds, which stabilize the protein structure. This means that two proteins with the same length but different sequences may store different levels of potential energy—depending on which amino acids are present.
Configuration adds stereochemical precision to this identity. It refers to the fixed spatial arrangement of atoms that cannot be altered without breaking covalent bonds. In proteins, this includes features such as chirality and cis-trans isomerism. The fact that human proteins are built almost exclusively from L-amino acids, while D-amino acids are largely excluded from biosynthesis, highlights how configuration is critical for biological recognition. It enforces the correct three-dimensional framework for folding and function, anchoring the molecule’s potential energy in an irreversible structural logic.
Conformation, by contrast, reflects the molecule’s dynamic potential. It represents the spatial flexibility that arises from rotations around single bonds—allowing proteins to shift between various shapes without changing their core identity. These movements are central to biological function. Active sites may open and close, flexible loops may rearrange, and domains may reorient during catalysis or binding. Although conformational changes do not alter the underlying constitution or configuration, they dramatically affect the molecule’s thermodynamic state by redistributing potential energy and modulating entropy.5
Together, these three layers—constitution, configuration, and conformation—form a unified framework for understanding how structural design translates into thermodynamic behavior. As shown in
| CCC Dimension | What It Represents | Illustrative Examples | Role in Potential Energy |
|---|---|---|---|
| Constitution | The basic chemical structure of a molecule, including the types of atoms and their bonding pattern. | The amino acid sequence of a protein; presence of cysteine residues capable of forming disulfide bridges. | Defines the molecule’s foundational capacity to form stable structures and store energy. |
| Configuration | The fixed three-dimensional spatial arrangement of atoms that requires bond-breaking to change. | L- versus D-amino acids; cis/trans isomers in peptide chains. | Governs precise biological interactions and stereospecific recognition. |
| Conformation | The flexible three-dimensional shape of a molecule, arising from rotation around single bonds. | Switching between active and inactive protein forms; protein folding into native shape; α-helices, β-sheets. | Determines the dynamic aspect of energy storage and release through structural transitions. |
folding involves a shift in conformation while maintaining the same constitution and configuration, yet this alone is enough to alter the protein’s stability, flexibility, and interaction potential. The CCC framework not only clarifies how energy and entropy are managed in biomolecules—it offers a practical and conceptual bridge between structure and function. By revealing how potential energy is embedded in both rigidity and flexibility, CCC helps explain the delicate balance that governs protein behavior in health, adaptation, and disease.
Thermodynamic Duality of Proteins: Structure and Function
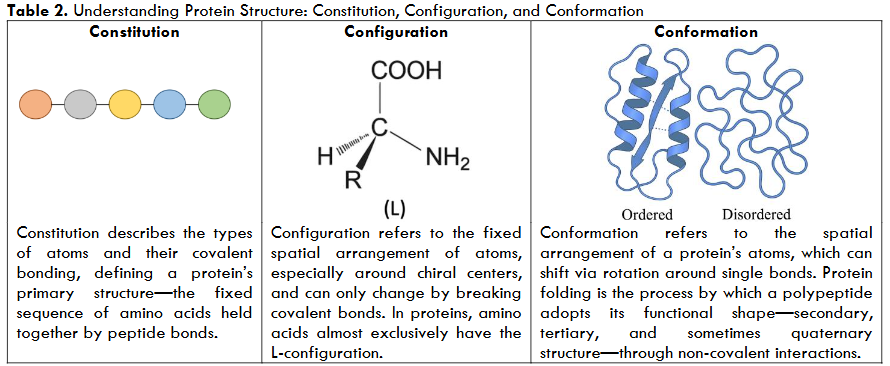
Proteins are dynamic molecular systems whose function emerges from a finely tuned thermodynamic balance between potential energy and entropy—a principle central to the CCC framework. As linear polymers of amino acids, proteins begin in a high-entropy, low-potential-energy state. Upon folding into ordered three-dimensional structures, they undergo a thermodynamic transition: potential energy increases due to structural constraint, while entropy decreases due to reduced conformational freedom.15
This transition is not merely geometric—it is deeply thermodynamic. In the unfolded state, the polypeptide chain explores a wide landscape of conformations. Folding limits this landscape, leading to entropic loss, yet introduces specific spatial arrangements that confer functional precision. Importantly, proteins do not fold into fully thermodynamically stabilized structures. Localized flexibility—often found in loops, side chains, and termini—preserves conformational entropy even within the native state. These flexible regions are critical for molecular recognition, catalysis, and allosteric regulation, underscoring how proteins maintain functionality through controlled disorder.16-17
The CCC framework helps explain this behavior. While constitution and configuration remain unchanged during folding, conformation provides the dynamic axis through which proteins navigate functional states. Transitions between conformers modulate potential energy and entropy without altering the molecule’s identity, allowing proteins to remain both stable and responsive.
Hydrophobic interactions play a crucial role in this folding process. As nonpolar residues bury within the protein core, they displace structured water molecules that previously formed cage-like hydration shells. The release of these water molecules into bulk solvent significantly increases the system’s entropy, partially compensating for the entropy lost in protein folding. In this sense, water acts as both an entropic reservoir and a molecular lubricant, facilitating folding, binding, and dynamic transitions.18 Each released water molecule significantly increases solvent entropy, contributing to the overall thermodynamic favorability.
Ligand binding shifts this equilibrium again. New interactions formed at the interface restrict conformational motion, raising potential energy and lowering internal entropy. However, the release of additional hydration water compensates through increased solvent entropy, contributing to the net thermodynamic favorability of binding. This redistribution—less internal entropy, more solvent entropy—reflects how proteins harness environmental interactions to maintain function.19-20
Ultimately, protein activity depends on achieving a thermodynamic middle ground. Excessive stabilization leads to rigidity and loss of adaptability, while unchecked flexibility invites misfolding and inefficiency. Function emerges not from static structure, but from the ability to oscillate within a controlled thermodynamic ensemble.21 The CCC framework highlights how this duality—between stored energy and accessible entropy—is embedded in protein architecture, and refined by evolution to support molecular precision, adaptability, and control.
Structural Proteins: Anchoring Potential through Rigidity
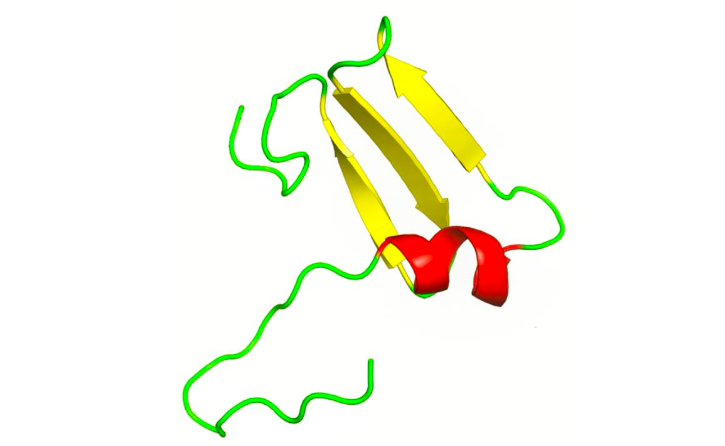
While biological systems harness the intricate flexibility and entropic potential of dynamic proteins, an equally vital aspect of molecular design is found in the thermodynamic stability and order of structural proteins. These remarkable molecules, including familiar examples like collagen, keratin, and elastin, form the very mechanical foundation of multicellular life. They are meticulously organized to provide the essential tensile strength, elasticity, and robust resistance to deformation required by tissues such as skin, tendons, cartilage, and hair, ensuring the integrity and function of complex biological systems.30 Their functionality emerges not from adaptability but from thermodynamic stability embedded in structural precision. Structural proteins like collagen and elastin are characterized by high degrees of organization, crucial for maintaining the integrity and functionality of various biological tissues throughout their life.31
Viewed through the lens of constitution, configuration, and conformation (CCC), structural proteins represent an optimized thermodynamic design for physical endurance. Constitutionally, they contain repetitive, highly ordered amino acid sequences—rich in glycine, proline, or cysteine—that promote polymerization and fiber formation.32 Configurationally, their atomic arrangements and stereochemistry are entropically restrictedly constrained, enabling dense packing, hydrogen bonding, and inter-chain crosslinking, which enhance their mechanical properties.33 Conformationally, they adopt thermodynamically stabilized, low-entropy structures—such as α-helices in keratin and triple helices in collagen—engineered to resist thermal fluctuations and mechanical distortion.
This conformational thermodynamic stability is not a deficiency but a functional imperative. Structural proteins do not require the conformational variability seen in enzymes or signaling complexes. Instead, their stored potential energy, encoded in constitution and locked into configuration, is stabilized—not mobilized—through structure. Their resistance to change is precisely what makes them reliable scaffolds in dynamic biological systems.34 Structural proteins’ unique design allows them to effectively dissipate mechanical stress and maintain long-term biological architecture over time, anchoring tissues against deformation.35
From a thermodynamic perspective, structural proteins represent a state of minimized entropy and maximal mechanical utility, maintaining stability without sacrificing structural fidelity.36 Unlike dynamic, catalytic proteins that rely on conformational flexibility, structural proteins exemplify how biological function can emerge not from motion but from purposeful immobility. Their role completes the functional spectrum of proteins governed by the interplay of potential energy and entropy, highlighting a critical balance between resilience and adaptability.

Misfolded Proteins: Origins and Implications
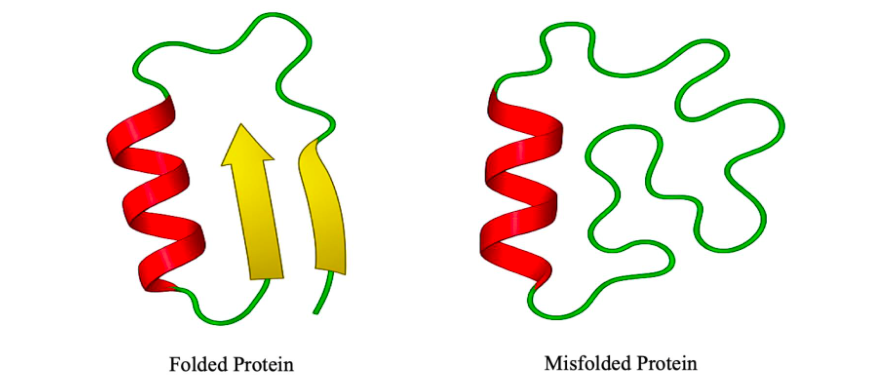
Protein misfolding emerges when the thermodynamic balance across the three structural layers—constitution, configuration, and conformation (CCC)—is disturbed. Under normal physiological conditions, a protein folds into its native structure by minimizing free energy through a coordinated process shaped by these three levels. Any deviation in this equilibrium may lead to the formation of misfolded states that are energetically misaligned and functionally compromised.37-38
At the constitutional level, the amino acid sequence defines the intrinsic potential energy landscape of the protein. Even minor alterations—such as single-point mutations—can significantly affect folding dynamics. Changes in side-chain chemistry may disrupt hydrophobic patterns, electrostatic balance, or steric compatibility, thereby redirecting the folding trajectory toward unstable intermediates. Translation errors or genetic mutations thus act as primary triggers for misfolding by reshaping the encoded folding code and its associated thermodynamic potential.39
At the configurational level, the spatial arrangement of atoms, particularly the stereochemical architecture, is normally fixed and stable. However, under pathological or stress-related conditions, configuration can be perturbed by irreversible modifications such as racemization, oxidation, or certain post-translational changes. Although rare, such events compromise the stereospecific interactions critical for initiating and stabilizing folding, leading to distorted folding pathways and misfolded end-products.40
Conformation, the most flexible and thermodynamic layer, is especially sensitive to environmental stress. Misfolded proteins often adopt non-native conformations rich in β-sheet content, which promote intermolecular hydrogen bonding and aggregation.41 While these conformations may appear energetically stable (enthalpically favored), they exhibit low conformational entropy and resist unfolding or refolding. This entropic constraint traps proteins in kinetically inaccessible states, contributing to the formation of insoluble aggregates like amyloid fibrils.42-43
Environmental insults such as oxidative stress, heat shock, or pH imbalance further exacerbate conformational instability by pushing proteins beyond their folding tolerance. Over time, these misfolded species accumulate, particularly when cellular quality control systems—chaperones,44 the ubiquitin–proteasome system, and autophagy—become overwhelmed or decline due to aging or chronic stress.45-46 The resulting proteostatic failure allows aggregates to persist, disrupt cellular functions, and trigger the pathogenesis of protein misfolding diseases, including Alzheimer’s, Parkinson’s, Huntington’s, and systemic amyloidosis.47-48
From a thermodynamic perspective, protein misfolding reflects a mismanagement of internal energy and entropy. Proper folding depends not only on reaching a low-energy state but also on maintaining sufficient entropy to explore and stabilize the correct conformational ensemble. When potential energy becomes trapped in non-functional conformations or entropy collapses prematurely, folding becomes directionless and inefficient. This breakdown in the thermodynamic logic embedded in the CCC framework underpins both the structural anomaly and the pathological consequences of misfolding.49
In this context, the CCC model offers a unified lens to understand how diverse molecular insults converge on a shared outcome: the destabilization of proteomic order through thermodynamic imbalance. Misfolding, therefore, is not merely a molecular malfunction—it is a failure of systemic structural logic, with wide-reaching consequences for cellular integrity and organismal health.
Balancing Reactivity and Stability in Drug Design
Drug–protein interactions are not static events but thermodynamic transitions governed by a molecule’s constitution, configuration, and conformation. Together, these structural layers determine how a drug distributes its potential energy and manages entropy as it binds and modulates a biological target. Effective drug design thus demands a careful arrangement of these forces to reshape the energy landscape of the protein with both precision and resilience.51
At the constitutional level, the drug’s atomic framework encodes the potential energy necessary for biological engagement. Central to this is the pharmacophore—a spatial arrangement of functional groups designed to interact selectively with the protein’s active or allosteric site. This region stores the potential energy needed to induce chemical or structural changes in the target. However, the pharmacophore alone cannot achieve specificity or adaptability without the support of structural features that contribute to the system’s entropy. This entropic flexibility arises from components such as linkers, side chains, and solvent-exposed termini. Thus, the drug’s architecture must be optimized to retain sufficient pre-binding entropy—to ensure adaptability and aqueous solubility—while not so flexible as to dilute interaction specificity or pharmacokinetic stability. An ideal design balances these forces, concentrating potential energy within the pharmacophore, while distributing entropy across modifiable structural features that facilitate dynamic recognition.52
When covalent bonds are formed with the protein, the drug’s constitution is irreversibly altered, releasing or redistributing potential energy to lock the protein in a new functional state. Such interactions demand exact positioning of reactive elements within the pharmacophore. In contrast, non-covalent binding relies on weaker but reversible forces, with hydrogen bond donors and acceptors playing a pivotal role. These features modulate local potential energy by forming directional contacts and displacing ordered water molecules, thereby improving binding through a favorable shift in solvent entropy.
Configuration adds another layer of thermodynamic complexity. A chiral drug interacting with a chiral protein does not simply form enantiomeric interactions, but diastereomeric complexes with distinct energetic and functional outcomes. One enantiomer may align perfectly with the protein’s active site, while its mirror image fails to engage or causes unintended effects. In some cases, the act of binding introduces new chiral axes, altering the drug–protein interface in structurally and energetically meaningful ways.53
At the conformational level, binding is accompanied by mutual structural adaptation. The drug may fold or twist into a lower-entropy state, while the protein may reorganize flexible regions into ordered motifs. This entropy loss must be compensated by sufficient gains in potential energy through precise contacts and favorable geometries. Drugs that cannot adopt compatible conformations—or fail to induce stabilizing changes in the protein—will have poor affinity or limited biological impact, regardless of their configuration or constitution.54
Viewed through the CCC framework, drug–protein binding is a multilayered thermodynamic negotiation. Constitution drives the energetic potential for interaction, configuration determines stereochemical compatibility, and conformation governs adaptability through entropy modulation. Ultimately, effective drug molecules are not passive ligands, but thermodynamic agents. They do not merely dock to a binding site—they reshape the target’s energetic profile, altering its function, its structure, and its role within the cellular network. The success of this interaction depends on how well the drug’s internal potential energy is channeled through the pharmacophore, how its entropy is managed through flexible linkers and dynamic regions, and how donor–acceptor motifs are employed to fine-tune the energetic cost of binding. Failures in drug design often stem from misalignments across these layers—whether due to excessive rigidity, incorrect chirality, or energetically incompatible conformers.55
Beyond Duality: Irreversible Binding, Stability, and Toxic Side Effects Despite major advances in drug development, many therapeutic candidates fail not due to lack of activity, but because of fundamental thermodynamic imbalances—specifically, the mismanagement of potential energy and entropy during interaction with biological systems. A drug’s efficacy is not only defined by its ability to bind strongly to a target, but also by how it modulates the dynamic equilibrium of that system without disrupting homeostasis. Failures in this balance often result in toxicity, resistance, or unintended systemic effects.56
Drugs that bind too tightly or irreversibly can appear potent in vitro, yet in vivo, they frequently impose excessive entropy loss, locking proteins into rigid conformations. These entropically restricted interactions impair biological adaptability, slow clearance, and increase the likelihood of off-target effects. Warfarin, a narrow therapeutic index anticoagulant, exemplifies this: its tight binding to plasma proteins makes it highly sensitive to dosing fluctuations, risking hemorrhage even from minor variations.57 Similarly, the LpxC inhibitor ACHN-975, developed for Gram-negative bacterial infections, was abandoned due to cardiovascular toxicity, caused by slow unbinding kinetics that disrupted physiological responsiveness.58
Beyond toxicity, entropically rigid binding intensifies selective pressure, accelerating the development of drug resistance. For instance, rifampicin, an antibiotic that binds tightly to bacterial RNA polymerase, often selects for rpoB mutations that reduce drug affinity and drive resistance.59 In HIV therapy, non-nucleoside reverse transcriptase inhibitors (NNRTIs) such as efavirenz face similar issues: point mutations in the enzyme’s allosteric site, driven by high-affinity, rigid binding, quickly render these drugs ineffective.60
These examples highlight a thermodynamic paradox: what begins as strong, seemingly effective binding becomes the very trigger for biological escape and treatment failure.61
From a thermodynamic perspective, reversible and entropy-aware interactions are favored. They maintain the protein’s conformational entropy, enabling it to respond dynamically to cellular conditions. Reversible binding supports regulated signaling, reduced toxicity, and mitigated selective pressure, especially in complex or adaptive diseases such as cancer or viral infections. In contrast, irreversible or ultra-stabilized binding traps energy within the drug–protein complex, narrowing therapeutic windows and destabilizing broader physiological systems.63
Understanding these outcomes through the constitution, configuration, and conformation (CCC) framework provides additional clarity. At the constitutional level, irreversible covalent inhibitors alter the drug and protein permanently, locking the system in a new energetic state. While useful in targeted interventions, this transformation must be applied with great caution due to the energetic irreversibility it imposes on the biological environment.64
The configurational layer—the three-dimensional spatial arrangement around chiral centers—is especially critical. A defining example is thalidomide, a drug introduced in the late 1950s as a sedative and anti-nausea treatment during pregnancy. Thalidomide exists as two enantiomers: one with sedative effects, and the other with teratogenic properties that disrupted embryonic development. The tragedy, which resulted in thousands of birth defects worldwide, arose from the failure to account for stereochemical configuration: although the drug was administered as a racemic mixture, in vivo interconversion between enantiomers occurred, rendering separation ineffective. This disaster underscores the fact that enantiomers, while sharing the same constitution, can generate drastically different biological outcomes—diastereomeric interactions with the chiral protein environments in the body may lead to distinct thermodynamic paths with divergent consequences.65
At the conformational level, drugs must retain enough flexibility to adapt to fluctuating protein structures. Overly rigid molecules may minimize entropy loss but often fail to engage with dynamic proteins or induce off-target effects due to structural incompatibility. Conformational adaptability is essential for productive binding, especially when targeting multi-state proteins or allosteric sites, where entropy plays a regulatory role. In sum, many therapeutic failures—whether due to toxicity, resistance, or stereochemical mismatch—are rooted in a breakdown of thermodynamic logic. The CCC framework highlights the need for constitutionally appropriate, configurationally precise, and conformationally adaptable drug designs.66-67
| Protein Class | Constitution | Configuration | Conformation | Entropy | Potential Energy | Primary Function |
|---|---|---|---|---|---|---|
| Dynamic Proteins (e.g., IDPs) | Defined amino acid sequence with flexible motifs | Correct stereochemistry and bonding | Highly flexible, sampling broad ensembles | High (adaptive flexibility) | High (latent, structurally embedded) | Signal transduction, regulation, context-sensitive binding |
| Folded Functional Proteins | Sequence optimized for specific tasks | Precise 3D atomic arrangement | Structured but locally flexible | Moderate (regulated motion) | Balanced (specific yet adaptable) | Enzymatic catalysis, receptor binding, molecular switching |
| Structural Proteins | Repetitive, information-dense sequences | Highly ordered and densely packed | Rigid, stable, resistant to conformational change | Low (constrained state) | High (stored as mechanical integrity) | Tissue scaffolding, mechanical support, architectural roles |
| Misfolded Proteins | May contain mutations or stress-induced changes | Disrupted stereochemistry or misaligned packing | Aberrant or aggregation-prone structures | High (non-productive) | Misdirected or lost | Pathological aggregation, disruption of proteostasis |
| Irreversibly Bound Complexes (e.g., covalent inhibitors) | Stable ligand or drug with strong target affinity | Often preorganized for tight interaction | Rigid, locked conformational state | Very low (restricted motion) | Very high (but poorly dissipated) | Irreversible inhibition; may cause resistance or toxicity |
Conclusion
This work presents a unified framework that reinterprets molecular and biological behavior through the dual lens of potential energy and entropy, anchored by the structural dimensions of constitution, configuration, and conformation. By revealing how proteins, whether dynamic, folded, structural, or misfolded, embody distinct thermodynamic signatures, we demonstrate that functionality is not a fixed trait of structural stability alone, but a dynamic expression of energy and flexibility. The concepts introduced here not only clarify long-standing biochemical phenomena such as protein folding and drug binding, but also offer a thermodynamically grounded path toward more adaptive and personalized therapeutic strategies. Recognizing entropy as a functional resource rather than a marker of disorder opens the door to a deeper understanding of health, disease, and recovery, positioning this duality as a foundational principle for future advances in precision medicine. We encourage future interdisciplinary efforts to empirically test this model and further integrate structural, computational, and thermodynamic data into a unified theory of protein behavior and therapeutic innovation.
Conflicts of interests:
None
Funding statement:
This research was made possible by the generous support of the Swiss Scientific Society for Developing Countries, to whom we extend our sincere thanks.
References
- Gonzalez D, Djulbegovic M, Antonietti M, Cordova M, Dayhoff G, Mattes R, et al. Intrinsic disorder in the human tear proteome. Invest Ophthalmol Vis Sci. 2023;64(11):14. https://doi.org/10.1167/iovs.64.11.14
- Alzeer J. Beyond disorder: A new perspective on entropy in chemistry. Am J Med Chem. 2024;5(1):1–5. https://doi.org/10.31487/j.ajmc.2024.01.01
- Csizmók V, Follis A, Kriwacki R, Forman‐Kay J. Dynamic protein interaction networks and new structural paradigms in signaling. Chem Rev. 2016;116(11):6424–6462. https://doi.org/10.1021/acs.chemrev.5b00548
- Hermann M, Hub J. SAXS-restrained ensemble simulations of intrinsically disordered proteins with commitment to the principle of maximum entropy. J Chem Theory Comput. 2019;15(9):5103–5115. https://doi.org/10.1021/acs.jctc.9b00338
- Alzeer J. Rethinking reactivity: How structure, energy, and entropy drive chemical transformations. J Pijar Mipa. 2025;20(4):590-597. https://doi.org/10.29303/jpm.v20i4.8795
- Váradi M, Vranken W, Guharoy M, Tompa P. Computational approaches for inferring the functions of intrinsically disordered proteins. Front Mol Biosci. 2015;2:45. https://doi.org/10.3389/fmolb.2015.00045
- Tenchov R, Zhou Q. Intrinsically disordered proteins: perspective on COVID-19 infection and drug discovery. ACS Infect Dis. 2022;8(3):422–432. https://doi.org/10.1021/acsinfecdis.2c00031
- Yang X, Zhou P, Shen S, Hu Q, Tian C, Xia A, et al. Entropy drives the ligand recognition in G-protein-coupled receptor subtypes. Proc Natl Acad Sci U S A. 2024;121(30):e2401091121. https://doi.org/10.1073/pnas.2401091121
- Fernández A, Fraser C, Scott LR. Purposely engineered drug-target mismatches for entropy-based drug optimization. Trends Biotechnol. 2012;30(1):1–7. https://doi.org/10.1016/j.tibtech.2011.07.003
- Huggins DJ, Sherman W, Tidor B. Rational approaches to improving selectivity in drug design. J Med Chem. 2012;55(4):1424–1444. https://doi.org/10.1021/jm2010332
- Bianchi G, Longhi S, Grandori R, Brocca S. Relevance of electrostatic charges in compactness, aggregation, and phase separation of intrinsically disordered proteins. Int J Mol Sci. 2020;21(17):6208. https://doi.org/10.3390/ijms21176208
- Sun C, Feng Y, Fan G. IDPsBind: A repository of binding sites for intrinsically disordered protein complexes with known 3D structures. BMC Mol Cell Biol. 2022;23(1):1. https://doi.org/10.1186/s12860-022-00434-5
- Alzeer J. Directionality of chemical reaction and spontaneity of biological process in the context of entropy. Int J Regen Med. 2022;5(2):1–7. https://doi.org/10.31487/j.rgm.2022.02.06
- Alzeer J. Exploring the dynamics of nucleophilic substitution reactions: Understanding the role of entropy and potential energy in SN1 and SN2 pathways. Am J Med Chem. 2023;4(1):1–4. https://doi.org/10.31487/j.ajmc.2023.01.02
- Chalikian T. Hydrophobic tendencies of polar groups as a major force in molecular recognition. Biopolymers. 2003;70(4):492–496. https://doi.org/10.1002/bip.10538
- Papaleo E, Saladino G, Lambrughi M, Lindorff‐Larsen K, Gervasio F, Nussinov R. The role of protein loops and linkers in conformational dynamics and allostery. Chem Rev. 2016;116(11):6391–6423. https://doi.org/10.1021/acs.chemrev.5b00623
- Campbell E, Kaltenbach M, Correy G, Carr P, Porebski B, Livingstone E, et al. The role of protein dynamics in the evolution of new enzyme function. Nat Chem Biol. 2016;12(11):944–950. https://doi.org/10.1038/nchembio.2175
- Dhusia K, Su Z, Wu Y. Understanding the impacts of conformational dynamics on the regulation of protein–protein association by a multiscale simulation method. J Chem Theory Comput. 2020;16(8):5323–5333. https://doi.org/10.1021/acs.jctc.0c00439
- Bellissent-Funel MC, Hassanali A, Havenith M, Henchman R, Pohl P, Sterpone F, et al. Water determines the structure and dynamics of proteins. Chem Rev. 2016;116(13):7673–7697. https://doi.org/10.1021/acs.chemrev.5b00664
- Deng N, Zhang P, Cieplak P, Lai L. Elucidating the energetics of entropically driven protein–ligand association: Calculations of absolute binding free energy and entropy. J Phys Chem B. 2011;115(41):11902–11910. https://doi.org/10.1021/jp204047b
- Strub C, Alies C, Lougarre A, Ladurantie C, Czaplicki J, Fournier D. Mutation of exposed hydrophobic amino acids to arginine to increase protein stability. BMC Biochem. 2004;5(1):9. https://doi.org/10.1186/1471-2091-5-9
- Dasgupta B, Tiwari S. Explicit versus implicit consideration of binding partners in protein–protein complex to elucidate intrinsic dynamics. Biophys Rev. 2022;14(6):1379–1392. https://doi.org/10.1007/s12551-022-01026-5
- Bah A, Vernon R, Siddiqui Z, Krzeminski M, Muhandiram R, Zhao C, et al. Folding of an intrinsically disordered protein by phosphorylation as a regulatory switch. Nature. 2014;519(7541):106–109. https://doi.org/10.1038/nature13999
- Uversky V, Dunker A. Understanding protein non-folding. Biochim Biophys Acta Proteins Proteom. 2010;1804(6):1231–1264. https://doi.org/10.1016/j.bbapap.2010.01.017
- Uversky V. Functional roles of transiently and intrinsically disordered regions within proteins. FEBS J. 2015;282(7):1182–1189. https://doi.org/10.1111/febs.13202
- Deiana A, Forcelloni S, Porrello A, Giansanti A. Intrinsically disordered proteins and structured proteins with intrinsically disordered regions have different functional roles in the cell. PLoS One. 2019;14(8):e0217889. https://doi.org/10.1371/journal.pone.0217889.
- DeForte S, Uversky V. Resolving the ambiguity: making sense of intrinsic disorder when PDB structures disagree. Protein Sci. 2016;25(3):676-688. https://doi.org/10.1002/pro.2864.
- Korneta I, Bujnicki J. Intrinsic disorder in the human spliceosomal proteome. PLoS Comput Biol. 2012;8(8):e1002641. https://doi.org/10.1371/journal.pcbi.1002641.
- Sotomayor-Pérez A, Ladant D, Chenal A. Calcium-induced folding of intrinsically disordered repeat-in-toxin (RTX) motifs via changes of protein charges and oligomerization states. J Biol Chem. 2011;286(19):16997-17004. https://doi.org/10.1074/jbc.M110.210393.
- Miranda-Nieves D, Chaikof E. Collagen and elastin biomaterials for the fabrication of engineered living tissues. ACS Biomater Sci Eng. 2016;3(5):694-711. https://doi.org/10.1021/acsbiomaterials.6b00250.
- Shoulders MD, Raines RT. Collagen structure and stability. Annu Rev Biochem. 2009;78:929-958. https://doi.org/10.1146/annurev.biochem.77.032207.120833.
- Dalla Valle L, Nardi A, Alibardi L. Isolation of a new class of cysteine-glycine-proline-rich beta-proteins (beta-keratins) and their expression in snake epidermis. J Anat. 2010;216(3):356-367. https://doi.org/10.1111/j.1469-7580.2009.01192.x.
- Nguyen T, Bashur C, Kishore V. Impact of elastin incorporation into electrochemically aligned collagen fibers on mechanical properties and smooth muscle cell phenotype. Biomed Mater. 2016;11(2):025008. https://doi.org/10.1088/1748-6041/11/2/025008.
- Casey D, Jawde S, Herrmann J, Mori V, Mahoney J, Suki B, et al. Percolation of collagen stress in a random network model of the alveolar wall. Sci Rep. 2021;11(1):95911. https://doi.org/10.1038/s41598-021-95911-w.
- Henninger H, Valdez W, Scott S, Weiss J. Elastin governs the mechanical response of medial collateral ligament under shear and transverse tensile loading. Acta Biomater. 2015;25:304-312. https://doi.org/10.1016/j.actbio.2015.07.011.
- Munkhuu B, Erdene L, Bayarsukh Z, Altantulga E, Baltsukh O, Tudev G, et al. Antioxidant and antihypertensive activity of collagen and elastin hydrolysate at different molecular weights. Adv Health Care Patient Saf. 2021. https://doi.org/10.2991/ahcps.k.211004.004.
- Khan AN, Khan RH. Protein misfolding and related human diseases: a comprehensive review of toxicity, proteins involved, and current therapeutic strategies. Int J Biol Macromol. 2022;223(Pt A):143-160. https://doi.org/10.1016/j.ijbiomac.2022.11.031.
- Quan N, Eguchi Y, Geiler-Samerotte K. Intra-FCY1: a novel system to identify mutations that cause protein misfolding. Front Genet. 2023;14:1198203. https://doi.org/10.3389/fgene.2023.1198203.
- Morales R, Moreno-González I, Soto C. Cross-seeding of misfolded proteins: implications for etiology and pathogenesis of protein misfolding diseases. PLoS Pathog. 2013;9(9):e1003537. https://doi.org/10.1371/journal.ppat.1003537.
- Ajmal MR. Protein misfolding and aggregation in proteinopathies: causes, mechanism and cellular response. Dis (Basel). 2023;11(1):30. https://doi.org/10.3390/diseases11010030.
- Ashraf GM, Greig NH, Khan TA, Hassan I, Tabrez S, Shakil S, et al. Protein misfolding and aggregation in Alzheimer’s disease and type 2 diabetes mellitus. CNS Neurol Disord Drug Targets. 2014;13(7):1280-1293. https://doi.org/10.2174/1871527313666140917095514.
- Morales R, Estrada L, Díaz-Espinoza R, Morales-Scheihing D, Jara M, Castilla J, et al. Molecular cross talk between misfolded proteins in animal models of Alzheimer’s and prion diseases. J Neurosci. 2010;30(13):4528-4535. https://doi.org/10.1523/jneurosci.5924-09.2010.
- Amm I, Sommer T, Wolf D. Protein quality control and elimination of protein waste: the role of the ubiquitin–proteasome system. Biochim Biophys Acta Mol Cell Res. 2014;1843(1):182-196. https://doi.org/10.1016/j.bbamcr.2013.06.031.
- Voisine C, Pedersen J, Morimoto R. Chaperone networks: tipping the balance in protein folding diseases. Nat Rev Mol Cell Biol. 2010. (Full citation or DOI not available).
- Samant R, Livingston C, Sontag E, Frydman J. Distinct proteostasis circuits cooperate in nuclear and cytoplasmic protein quality control. Nature. 2018;563(7731):407-411. https://doi.org/10.1038/s41586-018-0678-x.
- Guo X, Liu Y, Gao X, Kinoshita T, Fujita M. Calnexin mediates the maturation of GPI-anchors through ER retention. J Biol Chem. 2020;295(48):16393-16410. https://doi.org/10.1074/jbc.RA120.015577.
- Valastyan J, Lindquist S. Mechanisms of protein-folding diseases at a glance. Dis Model Mech. 2014;7(1):9-14. https://doi.org/10.1242/dmm.013474.
- Gidalevitz T, Kikis E, Morimoto R. A cellular perspective on conformational disease: the role of genetic background and proteostasis networks. Curr Opin Struct Biol. 2010;20(1):23-32. https://doi.org/10.1016/j.sbi.2009.11.001.
- Alzeer J. Halalopathy: role of entropy in the aging process. Am J Biomed Sci Res. 2022;16(2):147–154. https://doi.org/10.31487/j.ajmc.2023.01.02
- Fay A, Glickman M. An essential nonredundant role for mycobacterial DnaK in native protein folding. PLoS Genet. 2014;10(7):e1004516. https://doi.org/10.1371/journal.pgen.1004516.
- Claveria‐Gimeno R, Vega S, Abián O, Velázquez‐Campoy A. A look at ligand binding thermodynamics in drug discovery. Expert Opin Drug Discov. 2017;12(4):363-377. https://doi.org/10.1080/17460441.2017.1297418.
- Polyansky A, Zubac R, Žagrović B. Estimation of conformational entropy in protein–ligand interactions: a computational perspective. Methods Mol Biol. 2011;327-353. https://doi.org/10.1007/978-1-61779-465-0_21.
- Summa C, Langford D, Dinshaw S, Webb J, Rick S. Calculations of absolute free energies, enthalpies, and entropies for drug binding. J Chem Theory Comput. 2024;20(7):2812-2819. https://doi.org/10.1021/acs.jctc.4c00057.
- Sankar K, Jia K, Jernigan R. Knowledge-based entropies improve the identification of native protein structures. Proc Natl Acad Sci U S A. 2017;114(11):2928-2933. https://doi.org/10.1073/pnas.1613331114.
- Alzeer J. Halalopathy: stimulation of the immune system through enrichment of potential energy. Int J Regen Med. 2022;1–5. https://doi.org/10.31487/j.rgm.2022.01.02
- Alzeer J. Lifestylopathy as personalized medicine: a holistic approach to health. Med Res Arch. 2025;13(1). https://doi.org/10.18103/mra.v13i1.6209
- Rizzuti B, Bartucci R, Pey AL, Guzzi R. Warfarin increases thermal resistance of albumin through stabilization of the protein lobe that includes its binding site. Arch Biochem Biophys. 2019;676:108123. https://doi.org/10.1016/j.abb.2019.108123.
- Kalinin DV, Holl R. LpxC inhibitors: a patent review (2010–2016). Expert Opin Ther Pat. 2017;27(11):1227-1250. https://doi.org/10.1080/13543776.2017.1360282.
- Kong H, Byun J. Nucleic acid aptamers: new methods for selection, stabilization, and application in biomedical science. Biomol Ther (Seoul). 2013;21(6):423-434. https://doi.org/10.4062/biomolther.2013.085.
- Patel Y, Soni V, Rhee KY, Helmann JD. Mutations in rpoB that confer rifampicin resistance can alter levels of peptidoglycan precursors and affect β-lactam susceptibility. mBio. 2023;14(2):e0316822. https://doi.org/10.1128/mbio.03168-22.
- Andricopulo A, Guido R, Trivella D, Polikarpov I, Leitão A, Montanari C. Recent trends in structure-based drug design and energetics. Curr Protoc Bioinformatics. 2010;685-724. https://doi.org/10.1002/0471266949.bmc141.
- Gates A, Correia R, Wang X, Rocha L. The effective graph reveals redundancy, canalization, and control pathways in biochemical regulation and signaling. Proc Natl Acad Sci U S A. 2021;118(12):e2022598118. https://doi.org/10.1073/pnas.2022598118.
- Alzeer J. The role of buffers in establishing a balance of homeostasis and maintaining health. Am J Med Chem. 2023;4(1):1-6. https://doi.org/10.31487/j.AJMC.2023.01.01.
- Tokunaga E, Yamamoto T, Ito E, Shibata N. Understanding the Thalidomide Chirality in Biological Processes by the Self-disproportionation of Enantiomers. Sci Rep. 2018;8(1):17131. https://doi.org/10.1038/s41598-018-35457-6
- Varshney P, Sharma V, Yadav D, Kumar Y, Singh A, Kagithala N, et al. The impacts and changes related to the cancer drug resistance mechanism. Curr Drug Metab. 2023;24(12):787-802. https://doi.org/10.2174/0113892002266408231207150547.
- Gu Q, Zhu X, Yu Y, Jiang T, Pan Z, Ma J, et al. Type II and IV toxin-antitoxin systems coordinately stabilize the integrative and conjugative element of the ICESa2603 family conferring multiple drug resistance in Streptococcus suis. PLoS Pathog. 2024;20(4):e1012169. https://doi.org/10.1371/journal.ppat.1012169.