






Osteosarcoma: Advances in Models and Experimental Therapies
Osteosarcoma: A comprehensive review of model systems and experimental therapies
I. ABSTRACT
Osteosarcoma (OSA) is a highly malignant bone tumor for which more than 50% of patients have or will develop metastatic disease, resulting in an abysmal 5-year survival rate of <29%. Despite the advances in science and medicine, the etiology of OSA remains unclear. Similarly, the standard of care (surgery and chemotherapy) has changed little in the past 5 decades. This stagnation in treatment options is in part due to inadequate preclinical models for OSA; many of these models are oversimplified and do not account for the complexities of patient disease. Further, current treatments are harsh and invasive (e.g. high dose chemotherapy and potential limb removal) leading to a reduction in a patient’s quality of life (e.g. hearing loss, infertility, neuropathy), highlighting a need for developing more effective treatment strategies. Many experimental therapies have been tested in the preclinical and preclinical setting, with varying degrees of success. In this review, we will focus on pediatric and adolescent OSA, highlighting current animal models and experimental therapies.
II. Introduction
The term “osteosarcoma” was first coined by French surgeon, Alexis Boyer, in 1805 to describe osseous tumors he observed during his practice¹². Research focused on the cellular origins of OSA have identified osteoblasts, bone-forming progenitor cells, as a population of cells that give rise to OSA³. Though osteoblasts are a common cell of origin for OSA, the disease has a high degree of heterogeneity not only at the molecular level but also at the clinical level. Observations in the disproportionate presentation of OSA in the patient population have been correlated with skeletal structure size, which in part explains higher occurrences in males to females 1.2:1 and people of African origin compared to other ethnicities⁴⁵. Further, age is a large risk factor for OSA formation. OSA has a bimodal distribution with 87% of cases during adolescence (0–24 years of age with 8.6 cases per million) and a second, smaller peak in elderly populations (>60 years of age with 2 cases per million), which only accounts for 13% of all osteosarcoma cases⁵–¹⁰.
Despite the many risk factors for developing OSA, this cancer develops fairly predictably in children and adolescents with the most common site being near the metaphyseal growth plates of long bones of the limbs: femur and tibia at 42% and 19%, respectively, with 75% of tumors in the femur occurring distally and 80% of tumors in the tibia occurring proximally⁶⁸. The location and size of the primary tumor highly impacts outcome. Patients with tumors located on the axial skeleton, femur, or the trunk of the body have a decreased survival time of 10 years compared to patients with tumors located in the appendicular skeleton (e.g. humerus and tibia)¹¹¹².
The current standard of care for OSA, which has remained unchanged since the 1970s (neoadjuvant chemotherapy, tumor resection, adjuvant chemotherapy) is harsh and invasive with a 5-year survival of ~68%⁷⁸¹³. Current surgical procedures for OSA favor limb salvage, a highly invasive surgery to remove the tumor in an effort to preserve the major functions of the limb and carries high risks of mobility impairment and nerve damage. In cases where the limb cannot be saved, amputation—the previous gold standard, is performed to remove the tumor¹³¹⁴. However, the efficacy of these approaches drastically declines based on the size of tumors. Bulkier tumors (>15 cm diameter) have a 3.4 times greater morbidity risk due to challenges of limb salvage surgery, poorer response to chemotherapy, and greater probability of recurrence¹¹. Overall, males are less responsive to chemotherapy with a higher tendency of recurrence while females correlate with better response to chemotherapy having an overall greater survival of about 16 months¹¹¹⁵¹⁶. The highest rates of morbidity are attributed to the presence of metastatic disease. Metastasis of OSA occurs in the lung in >90% of documented cases while the next most common site of metastasis being other bones in 5–10% of documented cases¹⁷¹⁸. At initial presentation, overt metastases are detected in ~25% of patients, which is correlated to poor prognosis⁴. Unfortunately, most patients are presumed to have subclinical micrometastatic lesions at the time of diagnosis¹⁹. Genetic drivers and predisposition syndromes to OSA have been reviewed extensively by Beird et al¹⁰.
Despite advances in our understanding of OSA tumor biology, patient outcome remains poor, particularly with the presence of metastatic disease. Novel experimental treatments utilizing immunotherapeutic agents is a promising field of research with the potential to provide non-invasive and specific treatments for OSA. These treatments are highlighted in Figure 1. In this review, we will specifically focus on highlighting model systems and experimental therapies for the treatment of pediatric OSA.
Figure 1. Experimental immunotherapies for cancer. A schematic detailing current common immunotherapy methods used to treat patients with cancer.
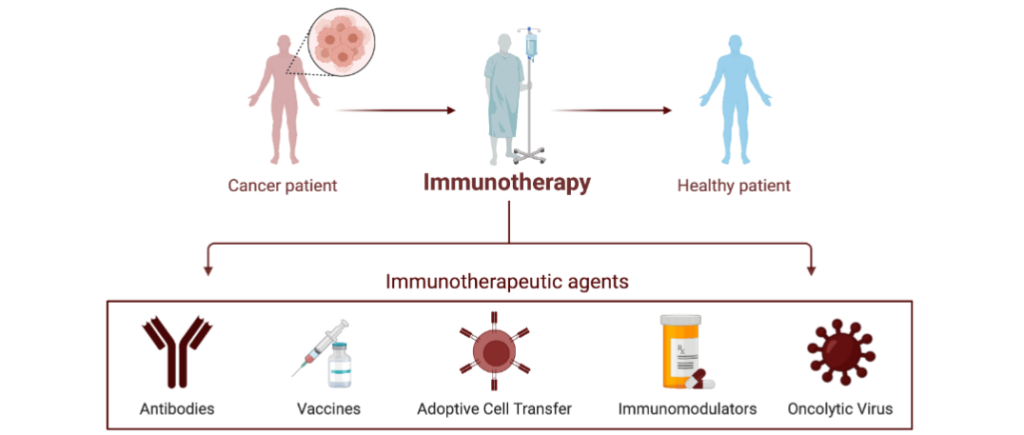
Cancer patient → Immunotherapy → Healthy patient
Immunotherapeutic agents:
- Antibodies
- Vaccines
- Adoptive Cell Transfer
- Immunomodulators
- Oncolytic Virus
III. In vitro and in vivo Model Systems
In vitro based tumor models range in their complexity and are used to provide insight into tumor genetics, growth/proliferation, migration/invasion, and response to experimental therapies. As we advance our understanding of tumor cell biology, the complexity of these models has developed rapidly. In vitro based models include cell line-based models, induced pluripotent stem cell-derived models, and three dimensional (3D) models. In vitro models have been summarized in table 1.
Table 1. In vitro models of osteosarcoma
Cell line based
Methods / Relevant Findings:
Adherent 2D cultures
Monolayer
Cell lines used include 143B, HOS, U2OS, SJSA-1, and G-292, among others
Advantages:
Widely established
Low-cost maintenance
Ease of culturing
Usability in various functional assays
Disadvantages:
Do not recapitulate the natural structure or microenvironment seen in patient tumors
Cannot mimic complex interactions between tumor cells, other cell types, and stimuli found in the tumor microenvironment
Altered response to experimental therapies
Changes in morphology, gene expression, signaling, polarity, topology, and loss of diverse phenotypes compared to original tumor tissue
References: 20,21
Induced pluripotent stem cell-derived
Methods / Relevant Findings:
TP53 and RB1 focused models
Rothmund-Thomson syndrome (RTS) model showed link to elevated mitochondrial respiratory complex I function
Advantages:
Able to be maintained indefinitely in an undifferentiated, pluripotent state using Yamanaka factors (OCT3/4, SOX2, KLF4, cMYC)
Ability to be differentiated into any human cell type
Fast to generate
Disadvantages:
Limited success generating these models from malignant cells
Engineering non-malignant cells to exhibit the genomic instability of OSA has not yet been achieved
Not well understood how iPSC models mimic tumor heterogeneity
Directed differentiation can result in a more immature phenotype
References: 22–30
Three dimensional (3D)
Methods / Relevant Findings:
Multicellular tumor spheroids (MCTSs)
Aggregates of cells grown in a 3D matrix that mimic the physical and biochemical properties of a tumor
Beyond MCTSs, 3D tumor bioprinting allows for the combination of cells, biomolecules, and biomaterials into organized, complex structures that mimic the characteristics of bone
Several MCTS and bioprinting models have been developed to study OS
Advantages:
Able to recapitulate critical elements of the tumor microenvironment, including cell-cell interactions
Sustain oxygen and nutrient gradients that result in necrotic cores as seen in many tumor types
Protein and gene expression profiles mirror patient data more closely than 2D models
Disadvantages:
Expensive
Time consuming to generate
3D modeling is in its relative infancy with more limited publications
References: 31–44
Animal models are a useful tool to understand the genetic basis of OSA and, more importantly, to advance preclinical studies for the generation of new therapeutic approaches. Animal models that accurately recapitulate the natural course of disease are the most informative, however, etiology and pathogenesis of OSA remain poorly understood. Therefore, establishment and induction of representative animal models remains challenging and incomplete. To date, mice are the most used species to generate OSA models, however, spontaneous cases of OSA in canines represent an additional relevant and validated model of OSA. These animal models are outlined in table 2.
Table 2. Methods of establishing animal models
Different in vivo model systems are listed with accompanying descriptions, immune competency of these models, as well as metastatic potential. All included methods for generating in vivo models have metastatic potential, however, some models may be transgene or cell line dependent.
Transgenics
Description:
De novo formation is induced by Cre-loxP-mediated inactivation of TSG alleles and/or activation of conditional oncogenes.
Tissue specific expression of the Cre-recombinase is achieved through crossbreeding with Cre transgenic mice, tamoxifen-inducible Cre-ERT transgenic mice, or by local administration of Cre-encoding lenti- or adenoviruses
Immune Component: Yes
Metastatic: Yes
References: 45
Allografts
Description:
LM8 derived by collecting OSA lung metastases derived from the Dunn-cell line-derived model
K7M2 derived from collecting lung metastases following intraosseous injection of K7 cell line (spontaneous murine OS)
Immune Component: Yes
Metastatic: Yes
References: 46–48
Xenografts
Description:
143B is derived from a 13-year-old patient’s tumor
HOSa is derived from a 13-year-old primary patient’s tumor. Has mutations in CDKN2A and TP53
U2OSA is derived from a 15-year-old patient’s primary. No mutations detected in 64+ commonly mutated genes
SaOS-2 is derived from an 11-year-old patient’s primary tumor. Has mutations in TP53 and RB1. Expresses TGF-beta type 1 and 2
MG-63 is derived from a 14-year-old patient’s tumor. Expresses high levels of TGF-beta
Immune Component: No
Metastatic: Yes
References: 49–53
PDXs
Description:
Generated using immunocompromised mice implanted subcutaneously with patient derived tissue
Immune Component: No
Metastatic: Yes
References: 54
Canine
Description:
Greater incidence in large and giant breed dogs, including Rottweilers, German Shepherds, Boxers, and Dobermans
Most frequent in middle-aged dogs between the ages of 6–10, however, smaller secondary peak in dogs between the ages of 1–2
Similar genetics to human patients; canine OSA is not inherited
Immune Component: Yes
Metastatic: Yes
References: 55,56
IV. Experimental Therapies
GENE THERAPY
Gene therapy is broadly defined as the transfer of genetic materials (DNA, RNA) into cells for the treatment of disease⁷. The genomic complexity and high mutational burden observed in OSA tumors has made gene therapies an attractive approach for prevention and treatment⁸.
Early attempts to treat OSA using gene therapy-based approaches focused on restoring function to commonly lost tumor suppressor genes (namely, TP53 and RB). TP53 is a multifunctional protein effectively involved in all of the hallmarks of cancer and has been found to be mutated in 9.5% of OSA patients⁵⁹⁶⁰. Phelan et al showed that delivery of a functional TP53 using the herpes protein VP22 resulted in OSA cell lines regaining the ability to induce apoptosis⁶¹. Densmore et al evaluated a murine OSA metastasis model with the treatment of P53 plasmid DNA, which showed a significant decrease in the number of tumor nodules and the size of the nodules that did form⁶². Several studies have shown that restoring or overexpressing P53 results in an increased sensitivity to chemotherapy drugs⁶³–⁶⁶.
RB is involved in cell cycle control by binding E2F family transcription factors until being phosphorylated by the CDK4/cyclin-D complex and is inactivated in ~50% of OSA tumors⁶⁷⁶⁸. Despite the frequency of RB mutations in OSA patients, RB heterozygous mice do not develop OSA and RB knockout (KO) is lethal⁶⁹⁷¹. While RB mutations do not appear to be causative of OSA, loss of heterozygosity (LOH) at the RB locus is common in high-grade tumors and is associated with poor patient prognosis⁷²⁷³. Several studies have aimed to restore function to the RB pathway using a replication-deficient recombinant adenovirus vector and showed a decrease in tumorigenicity in vivo⁷⁴⁷⁵. However, this approach was less effective in established xenograft models.
Beyond tumor suppressor genes, there are other genomic alterations that result in overexpression or upregulation; these oncogenes have been targeted for suppression for the treatment of OSA and are summarized in Table 3.
Table 3. Summarized gene therapy targets
Oncogenes targeted for suppression for the treatment of OSA with variable degrees of efficacy during in vitro and in vivo testing.
Vascular Endothelial Growth Factor (VEGF)
Findings in OS:
Pro-angiogenic factors associated with tumor angiogenesis
Silencing VEGF using siRNAs suppressed tumor growth, reduced angiogenesis, and downregulation of PI3K and AKT in vivo
References: 76–78
Apurinic/Apyrimidinic Endonuclease 1 (APE1)
Findings in OS:
Plays a critical role in DNA repair and redox regulation
APE1 has been shown to be overexpressed in OSA and is associated with chemoresistance and poor patient outcome
miRNAs silencing of APE1 demonstrated the function of APE1 in inhibiting DNA damage repair and sensitization of OSA cells to cisplatin
Silencing APE1 expression showed anti-angiogenic effects, increased apoptosis, and VEGF suppression in OSA xenografts
References: 79–83
Insulin-Like Growth Factor 1 (IGF1R)
Findings in OS:
14% of OSA tumors have amplification of IGF1R
miRNA targeting of IGF1R inhibited activation of AKT and ERK signaling pathways
References: 84,85
c-Jun
Findings in OS:
High grade OSAs show an increase in the transcription factor c-Jun
c-Jun DNAzyme inhibited OSA growth and metastasis in vitro and in vivo in an orthotopic OSA model
miRNA targeting of Hsp70 to modulate downstream JNK/JUN signaling pathway modulated OSA chemoresistance
References: 86–89
Ezrin
Findings in OS:
Metastasis-associated Ezrin functions in a cancer setting by allowing cells to overcome several stresses during the metastatic cascade, including initiating the translation of new proteins and efficient ATP generation
OSA patients with lung metastases have shown a fivefold increase in Ezrin mRNA expression when compared to non-metastatic patients
In vitro use of Ezrin-specific siRNA showed significant reduction in growth rate and cell morphology
References: 90–93
Cyclin A2 (CCNA2)
Findings in OS:
CCNA2 is a critical regulator of cell division that has been shown to be overexpressed in OSA and is linked to poor patient prognosis and metastasis
Knock down of CCNA2 using siRNAs showed dramatic decreases in proliferation in vitro
miRNA repression of CCNA2 inhibited proliferation, migration, and colony-formation of aggressive OSA cell lines
References: 94–97
Urokinase plasminogen activator and receptor (uPA/uPAR)
Findings in OS:
uPA and uPAR play critical roles in tumor invasion, migration, angiogenesis, and metastasis
Expression of uPA and uPAR have been reported in human and canine OSA with an association between enhanced metastatic behavior
Anti-uPAR DNAzymes showed a decrease in uPAR transcript by over 80%, resulting in significantly decreased OSA cell invasion in vitro
shRNA silencing of uPAR and small molecule inhibition of uPA showed decreased migratory response and decreased or total inhibition of metastasis of OSA in vitro and in vivo
References: 98–104
Several studies have utilized gene therapy approaches that are less dependent on patient tumor genetics to provide a more generalized therapy; approaches include introduction of IL-12, B7-1 gene transfer, herpes simplex thymidine kinase (HSV-TK), among others¹⁰⁵–¹⁰⁷. While in vivo results were promising, these models used OSA cells transduced prior to injection, which is not representative of treatment administered in a clinical setting. A major challenge for gene therapy-based treatments is that each OSA patient has unique genetics and mutational burdens. Despite some initial, promising success, a major challenge facing gene therapy-based precision medicine for OSA is that the most frequent genomic alterations (namely, TP53 and RB) are challenging to target therapeutically and in vivo success has not translated to the patient setting¹¹⁰. More universally applicable gene therapy approaches are critical to improving patient outcome, particularly for metastatic disease. One such target may be forkhead box P1 (FOXP1), which in OSA acts as an oncogene by suppressing the TP53-P21-RB cascade¹¹¹.
considerations to make gene therapy a more feasible approach to treat OSA include efforts to decrease the cost and time to develop these therapies.
SMALL MOLECULES INHIBITORS
Since the dawn of modern cell biology in the 1980s, small molecule inhibitors have become one of the primary methods for targeted cancer treatment¹¹²¹¹³. Small molecule inhibitors work by interrupting target proteins’ function by binding to these proteins or their receptors¹¹⁴. When compared to other cancer therapies, small molecule inhibitors have several advantages, including binding a wider range of targets due to their small size, ability to be taken orally, and ability to penetrate the blood-brain barrier¹¹⁵. The majority of small molecule inhibitors target protein kinases, which catalyze the transfer of the phosphate group in ATP to the hydroxyl group of substrate proteins¹¹⁶. Small molecule inhibitors tested for OSA are summarized in Table 4.
Table 4. Summarized small molecule inhibitors
Each small molecule has one or many targets with variable degrees of efficacy against osteosarcoma during preclinical and clinical testing.
Anlotinib
Targets: VEGFR1-3, PDGFRα/β, FGFR1-4, Aurora B, EMT, RET, KIT
Findings in OS: Suppresses tumor proliferation, angiogenesis, and metastasis in vitro and in vivo. Treatment increased chemo-sensitivity
References: 117–121
Apatinib
Targets: VEGFR1-2, KIT, RET, v-src avian sarcoma viral oncogene homolog
Findings in OS: Promotes apoptosis and autophagy while inhibiting invasion, migration, and PD-L1 expression in vitro. Phase II clinical trial showed a partial response rate of 43%
References: 122–125
Axitinib
Targets: VEGFR1-3, PDGFRα/β, RET, KIT, FGFR1, CSF-1R
Findings in OS: Children’s Oncology Group Phase I and pilot consortium trial showed 2 OSA patients presented with stable disease
References: 126–128
Cabozantinib
Targets: VEGFR1-3, RET, KIT, FLT3, MET, PDGFRα/β
Findings in OS: Decreases proliferation and migration in vitro by inhibiting ERK and AKT. Phase II clinical trial showed 12% of OSA patients had a partial response, 33% had 6-month stable disease
References: 129–132
Cediranib
Targets: VEGFR1-3, KIT, FGFR1, PDGFRα/β
Findings in OS: Showed no improvement in survival against OSA xenograft model. Phase I clinical trial showed 1 of 4 OSA patients had a partial response
References: 133–136
Dasatinib
Targets: BCR-ABL, SRC, LCK, YES, FYN, KIT, EPHA2, PDGFRβ
Findings in OS: Evaluated in canine OS; 2 dogs showed stable disease to partial remission
References: 137,138
Fruquintinib
Targets: VEGFR1-3
Findings in OS: Improvement in proliferation-free survival in retrospective study
References: 139,140
Imatinib
Targets: PDGFRα/β, RYK, EGFR, EPHA2, EPHA10, IGF1R, KIT, BCR-ABL
Findings in OS: Inhibited downstream signaling molecules such as AKT and ERK in vitro. Dose dependent anti-proliferative effect. This did not translate to in vivo mouse studies. Did not translate to the clinical setting
References: 141–145
Lenvatinib
Targets: VEGFR1-3, FGFR1-4, PDGFRα, RET
Findings in OS: No preclinical studies. Clinical trials have been underwhelming with few partial responders and a high incidence of serious treatment associated adverse events
References: 146–148
Nintedanib
Targets: VEGFR1-3, FGFR1-2, PDGFRα/β
Findings in OS: Increases apoptosis in vitro and blocked the formation of lung metastases in vivo
References: 149–151
Pazopanib
Targets: VEGFR1-3, PDGFRα/β, KIT
Findings in OS: In vivo combination treatment with chemotherapy drug topotecan reduced primary tumor growth. Retrospective study showed promising results of pazopanib treatment on 15 relapsed OSA patients
References: 152–154
Regorafenib
Targets: VEGFR1-3, PDGFRα/β, KIT, FGFR1-2, RET, TEK
Findings in OS: Treatment in vitro and in vivo induces apoptosis, reduces tumor growth, and reduces invasion. Findings did not translate to the clinical setting
References: 155–158
Sorafenib
Targets: AXL, RAF, FGFR2, KIT, PDGFRα/β, RET
Findings in OS: No effect of treatment on canine OSA cell viability in vitro. Phase II clinical trial had underwhelming results for patients with advanced or unresectable disease
References: 138,159,160
Sunitinib
Targets: VEGFR1-3, KIT, FLT3, AXL, EPHB2, FGFR2, IGF1R, RET
Findings in OS: Reduced tumor burden, tumor vasculature, and lung metastasis in mouse models of human OSA. Combination therapy even more effective
References: 161–165
Despite success in other tumor types and in vitro and in vivo OSA models, there are still many challenges facing small molecule inhibitors. Success has been limited and further investigation into safety, low response rates, patient-patient genetic variation, and tumor resistance is critical to making small molecule inhibitors more effective for the treatment of OSA.
IMMUNOTHERAPY
Since William Coley, the father of immunotherapy, first tried to harness the immune system in 19th century bone cancer patients, immune-based therapies have drastically transformed cancer research and patient care¹⁶⁶. Presently, cancer immunotherapies function by aiding the body in the detection and/or elimination of cancer cells¹⁶⁷. Immunotherapy based approaches to treat cancer include monoclonal antibodies, cancer vaccines, and adoptive cell therapy. The timeline of development, type, and target of immunotherapy for OSA is showcased in Figure 2.
MONOCLONAL ANTIBODIES
Antibodies are glycoproteins in the immunoglobulin superfamily that recognize and neutralize foreign antigens while initiating an immune response¹⁶⁸. Monoclonal antibodies (mAbs) are antibodies produced by individual B cell clones and specifically recognize a single antigenic determinant, or “epitope”¹⁶⁹. A single pathogen induces the production of numerous antibodies targeting different epitopes found on that pathogen. Targeted monoclonal antibodies induce tumor cell death through a variety of mechanisms, including blocking critical tumor cell receptors or ligands, antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), or complement dependent cytotoxicity (CDC)¹⁷⁰.
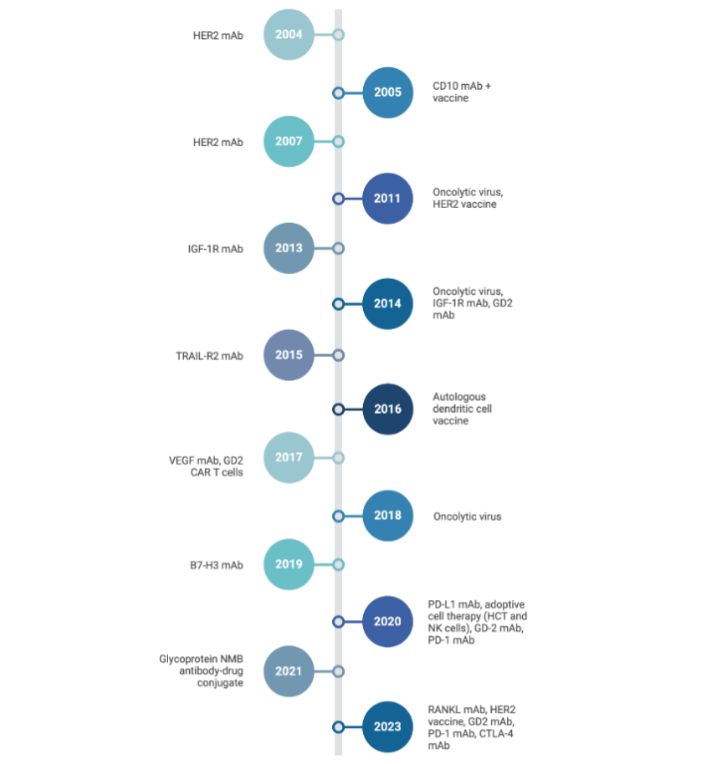
Figure 2. Timeline of osteosarcoma treatment and therapeutic development
Timeline showing development of immunotherapies for OSA:
Table 5. Summary of many monoclonal antibody treatments for osteosarcoma
Each monoclonal antibody therapy has a single target with variable degrees of efficacy against osteosarcoma during preclinical and clinical testing.
Bevacizumab
Target: VEGF
Mechanism and Findings in OS: Inhibit angiogenesis and thought to improve delivery of chemotherapy agents. Did not improve OSA patient outcome
References: 171–173
Ramucirumab
Target: VEGFR2
Mechanism and Findings in OS: Dominant VEGF receptor responsible for mediating the functions of VEGF. Underwhelming results in murine models of OS
References: 174,175
Nivolumab
Target: PD-1
Mechanism and Findings in OS: PD-L1/2 expression is negatively associated with patient outcome, recurrence, and metastasis in OS. In vivo treatment showed a decrease in the number of metastases, tumor apoptosis, decreased tumor cell proliferation, blockade of pSTAT3/ERK1/2 signaling, increased immune cell infiltration, and a decrease in pro-inflammatory M2 macrophages. Several clinical trials have been run to evaluate PD-1/PD-L1 blockade in OSA but limited success has been seen
References: 176–188
Ipilimumab
Target: CTLA-4
Mechanism and Findings in OS: CTLA-4 is expressed by OSA tumors and cell lines but preclinical investigations into CTLA-4 blockade in OSA are limited. Clinical trials targeting CTLA-4 have shown limited effect in OSA
References: 189–192
Anti-LRRC15
Target: LRRC15
Mechanism and Findings in OS: LRRC15 is known to be tumorigenic and is overexpressed in OSA. It is also associated with increased incidence of metastasis, chemoresistance, and reduced patient survival. OSA cells high in LRRC15 expression showed inhibited growth when treated with anti-LRRC15. PDX models showed tumor growth inhibition. Phase I clinical trial evaluating anti-LRRC15 found an overall patient response rate of 20%
References: 193–196
Trastuzumab
Target: HER2
Mechanism and Findings in OS: 40–80% of OSAs express HER2 to varying degrees, with ~30% showing high levels of HER2. Due to the limited nature of anti-HER2 preclinical testing in OSA, success in other tumor types prompted a phase II clinical trial of trastuzumab in OSA. The outcome was poor for all patients involved in the study
References: 197–199
Anti-EGFR
Target: EGFR
Mechanism and Findings in OS: EGFR has been found to be highly expressed in ~50% of OSA patients and while it is not a driver of tumor growth, it does contribute to chemoresistance. Treatment with anti-EGFR potentiated and directed NK cell activity toward OSA cells in vitro
References: 200–203
Teprotumumab
Target: IGF-1R
Mechanism and Findings in OS: IGF-1R is a glycoprotein overexpressed in OSA, aiding in tumor progression through transformation, proliferation, chemotherapy resistance, and metastasis. Preclinical testing showed promise in OSA with decreased tumor growth, increased event-free survival, and decreased AKT signaling. Phase I and II clinical trials evaluating cixutumumab (anti-IGF-1R mAb) in solid tumors showed limited efficacy
References: 204–207
Glembatumumab
Target: gpNMB
Mechanism and Findings in OS: Preclinical studies showed high gpNMB expression on OSA cell lines and potent anti-tumor effects. A phase II clinical trial looking at glembatumumab in 22 relapsed or refractory OSA patients showed only one patient with a partial response
References: 208,209
Anti-SEMA4C
Target: SEMA4C
Mechanism and Findings in OS: SEMA4C was shown to be overexpressed and antibody targeting promoted adhesion while reducing proliferation, colony formation, migration, wound healing, tumor growth, and metastasis in OSA
References: 210
Thus far, monoclonal antibodies have not provided a breakthrough in the treatment of OSA. Many monoclonal antibody treatments have shown underwhelming results in clinical trials with limited efficacy (Table 5). However, they have led to the understanding of many important pathways and tumor response mechanisms that will be critical in identifying cures. A preclinical study using the K7M2 model assessed a combination CTLA-4 and PD-1/PD-L1 blockade and showed promise and may help patients where PD-1/PD-L1 blockade alone is not sufficient due to tumor escape²¹¹. SEMA4D has emerged as another promising target in the semaphorin family, especially due to signaling through its receptors PLXNB1 and PLXNB2, which regulates cell migration, survival, and tumor vascularization²¹²¹³. Monoclonal antibodies may serve an important role in preventing metastatic disease, the primary indicator of poor patient outcome for OSA.
CANCER VACCINES
Immune escape is one of the hallmarks of cancer. To resensitize the immune system of a patient, cancer vaccines aim to stimulate anti-tumor immunity through the presentation of tumor antigens²¹⁴. One group used an attenuated Salmonella typhimurium vaccine to augment innate immune responses²¹⁵. They achieved a partial response in one of the four canine OSA patients treated, a modest effect. Hashii et al evaluated a vaccine targeting Wilms tumor gene 1 (WT1), which is overexpressed in many pediatric cancers²¹⁶. Unfortunately, the single OSA patient in this study did not benefit. Himoudi et al tested an autologous dendritic cell vaccination against OSA in a phase I clinical trial²¹⁷. Of the twelve OSA cases, only two patients showed strong, T-cell immune responses while another patient had a strong, but non-specific immune response. Finocchiaro et al evaluated the vaccination of canine OSA cell lines with a vaccine containing cytokine-producing cells (human GM-CSF and human IL-2)²¹⁸. Preclinical in vitro experiments showed promise and early clinical trial data shows a partial response in one canine patient and stable disease in two of the five canine patients that have undergone treatment. Gentschev et al showed potent effects of an oncolytic vaccinia virus against canine OSA in vitro²¹⁹. Recently, Cascini et al used a TLR9 agonist that effectively activated an innate and adaptive immune response, indicating this agonist acted as an in situ vaccine against OSA²²⁰. Mason et al have reported on a recombinant Listeria monocytogenes vaccine that expresses a chimeric human HER2/neu construct²²¹. In spontaneous canine OSA, this HER2 listeria vaccine inhibited lung metastasis and prolonged survival.
As we continue to explore novel therapies for OSA, it is becoming increasingly apparent that combination therapies are critical, raising a number of questions. How will cancer vaccines fit into this picture? Can we develop vaccines that will sensitize the tumor to the patient’s immune system to prevent metastatic disease or even sensitize the tumor to additional chemotherapy or adoptive cell therapy?
ADOPTIVE CELL THERAPY
Over the past few decades, harnessing the immune system has become an attractive area of research for the treatment of cancer. Although targeted immunotherapies, such as monoclonal antibodies and cancer vaccines, have been efficacious in improving survival in some tumor types, solid tumors remain challenging. Adoptive cell therapy is another immunotherapy that has gained significant attention and is rapidly evolving. This cell-based therapy involves isolating patient or healthy donor immune cells, expanding them with or without modifications, and then administering these to the patient to mount an antitumor response. There are three sources of cells currently being developed for use as cell-based therapies; these include autologous, allogeneic, and xenogeneic cells²²². Autologous cells are derived from the patient, allogeneic cells are human but from a healthy donor (not the patient), and xenogeneic cells are of non-human origin. Adoptive cell therapies can be grouped into three primary types, these include tumor-infiltrating lymphocytes (TILs), T cell receptor (TCR) therapy, and chimeric antigen receptors (CARs).
TUMOR-INFILTRATING LYMPHOCYTES
Tumor-infiltrating lymphocytes (TILs) are detected in >75% of OSAs and include T cells, B cells, NK cells, macrophage, and mast cells²²³. What makes TILs unique is they are considered to have higher specific reactivity against tumors when compared to normal lymphocytes²²⁴. Unfortunately, patient data suggests that OSA is largely an immunologically “cold” tumor that lacks tumor neoantigens and immune cell infiltration²²⁵. Traditionally, TILs are harvested from resected tumors, expanded in vitro, and then administered to patients²²⁶. As of recent years, TIL therapies only utilize T cells²²⁷. Casanova et al showed that in OSA, the presence of TILs was correlated with a better prognosis, supporting further investigation of TIL adoptive cell therapies for the treatment of OSA²²⁸. Zhou et al performed a retrospective analysis of adoptive TIL therapy plus the addition of anti-PD1 in patients with metastatic OSA²²⁹. They found that patients given this combination therapy exhibited increased progression free survival and overall survival. Wang et al reported on the successful isolation and expansion of TILs and the subsequent administration of these cells with anti-PD1 therapy²³⁰. Similar to the retrospective study, they found that the combination therapy of TILs and anti-PD1 resulted in improved patient outcome.
As is evident by the dearth of publications, strategies for TIL isolation from OSA and subsequent expansion are not yet optimized, resulting in inadequate cell numbers to be administered therapeutically. Another limitation of TIL based adoptive cell therapies is the immunosuppressive nature of OSA. OSA is known to support a very immunoregulatory and inhibitory microenvironment, which could prevent the activation of TILs²³¹.
T CELL RECEPTOR THERAPY
Building on the concepts of TILs, T cell receptor (TCR) therapy uses engineered T cells to express a novel TCR for the recognition of tumor associated antigen (TAA)³². T cells are typically transduced using viral based methods, however, non-viral systems, such as transposons and CRISPR/Cas9, are being developed²³³. Watanabe et al generated a TCR multimer with high avidity for naturally occurring TAAs on OSA cells²³⁴. While TCR therapies have shown promise in other tumor types, the field remains largely unexplored with respect to OSA. Due to the genomic instability and variability often seen between patients, identification of individual patient neoantigens will be cost and labor intensive. Another concern, especially with an immunologically cold tumor like OSA, is ensuring that these TCR engineered T cells traffic to the tumor. Further, a major limitation of all T cell-based therapies is that they are HLA restricted and thus must be HLA matched to be effective and safe for the patient. Success of TCR-based adoptive cell therapies will be largely dependent on improving peptide MHC-TCR interactions and identifying shared TAAs²³⁵.
CHIMERIC ANTIGEN RECEPTORS
CARs are engineered receptors designed to graft immune effector cells with the ability to specifically target TAAs independent of MHC restriction and activate modified cells through signal transduction²³⁶. Historically, most studies have evaluated CARs in T cells for the treatment of various cancers, however, other immune cell types are emerging as attractive effector cells, such as NK, macrophage, and others²³⁷. CAR-T cell therapies have been very successful in the treatment of hematological cancers, which has sparked interest in designing new CARs for the treatment of more diverse malignancies, including solid tumors²³⁷. CAR constructs are composed of an extracellular binding domain known as the single-chain fragment variable (scFv), a hinge region, transmembrane domain, intracellular signaling domains, and costimulatory domains²³⁸. Three CAR targets that have received notable attention for OSA include HER2, B7H3, and GD2. Ahmed et al evaluated HER2 CAR T cells against OSA, showing superior cytotoxicity in vitro and in vivo²³⁹. Rainusso et al showed similar results²⁴⁰. Talbot et al showed potent antitumor activity of B7H3 CAR T cells against OSA²⁴¹. Similarly, Zhang et al also evaluated B7H3 CAR T cells showing anti-tumor activity against OSA in vitro and in vivo²⁴². Interestingly, Hidalgo et al evaluated CAR T cells against, using B7H3 as a target and showed antitumor activity in vitro and in vivo²⁴³. Their strategy utilized intermediary switch molecules, which are adaptors that target TAAs and selectively bind the CAR, mediating interactions between CAR T cells and tumors allowing for improved safety. Chaloner et al generated GD2 CAR T cells, which showed effective killing of OSA in vitro²⁴⁴. Interestingly, OSA cells that survived treatment with GD2 CAR T cells upregulated PD-L1, suggesting that a combination therapy may be beneficial. Fernández et al evaluated NKG2D-CAR T cells against OSA, which showed increased cytotoxic activity in vitro and in vivo²⁴⁵. Huang et al tested interleukin (IL)-11Rα CAR T cells against OSA, showing a regression of pulmonary metastases in a mouse model²⁴⁶. Wang et al generated CD166 CAR T cells, which showed antigen expression dependent cytotoxicity against OSA in vitro and tumor regression in vivo²⁴⁷. CAR T cells targeting an isoform of alkaline phosphatase, ALPL-1, generated an effective anti-tumor response against OSA in vitro and in vivo²⁴⁸.
To date, there have been many targets of CAR T cell therapies, and many have made it into clinical trials (Figure 3). To further improve the success of CAR T cell therapies, particularly against solid tumors, there are several limitations to address. First, these therapies are associated with several toxicities in patients, including cytokine release syndrome (CRS), prolonged cytopenias, neurological toxicity, among others²⁴⁹. Other limitations include antigen escape, limited persistence, poor homing/infiltration, and inactivation by immunosuppressive tumor microenvironment²⁵⁰.
Therapeutic Targets
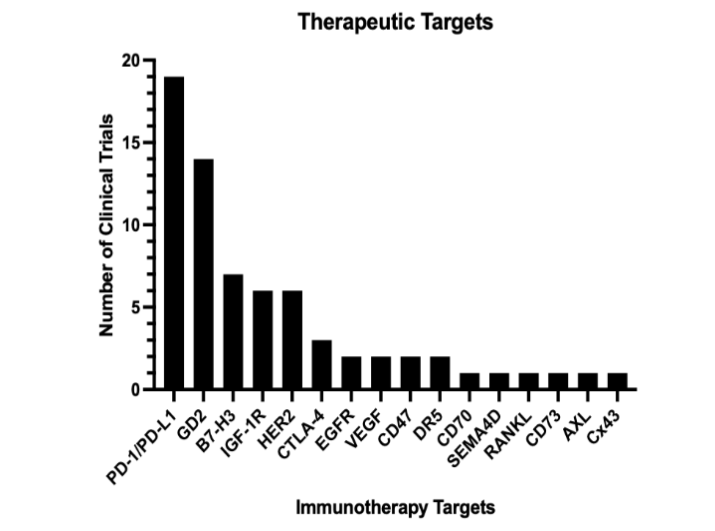
Figure 3. Osteosarcoma clinical trial therapeutic targets that have been tested.
The bar graph shows the frequency of different targets in completed clinical trials.
Immunotherapy Targets:
PD-1/PD-L1, GD2, B7-H3, IGF-1R, HER2, CTLA-4, EGFR, VEGF, CD47, DR5, CD70, SEMA4D, RANKL, CD73, AXL, CX43
Y-axis: Number of Clinical Trials (up to ~20) V.
V. Conclusion
Over the last several decades, scientific efforts have advanced our understanding of osteosarcomagenesis, its genetic landscape, metastasis, prognostic markers, and the development of effective preclinical in vitro and in vivo model systems. New molecular techniques have allowed for the generation and exploration of novel targets and therapies. Unfortunately, patient outcome has remained stagnant for five decades, with a dismal prognosis for patients with relapsed and/or metastatic disease. As a rare tumor, human OSA research would benefit from continued interdisciplinary research through collaboration with veterinarians working with canine patients, a species that sees more than 25,000 cases of OSA annually. The variation between patients with respect to tumor antigen expression poses another major challenge for developing an all-encompassing therapy. However, some targets are more broadly expressed by OSAs, such as HER2, B7H3, and GD2. OSAs with greater immune infiltration hold a better prognosis than their typical “cold” counterparts.
Cell-based therapies hold great promise in resensitizing these tumors to immune activity. Novel immunotherapy targets, such as Ephrin-A2²⁵¹, are critically needed. To date, all OSA CAR therapy publications have utilized T cells, however, many other immune cell types hold promise as potential effector cells, including natural killer (NK) cells, γ/δ T cells, and monocytes/macrophage. These other immune effector cells overcome several limitations of CAR T cells, notably patient toxicities. However, cell-based and other immunotherapies are limited by tumor immune escape, something commonly seen in OSA. Developing immunotherapies to target multiple antigens and/or expressing factors to promote their sustainability would undoubtedly enhance success in the treatment of OSA. Current targets of interest for immunotherapy against osteosarcoma are shown in Figure 4. Finally, combination therapy utilizing multiple targets will likely be critical to prolonged tumor control in relapsed and/or metastatic patients.
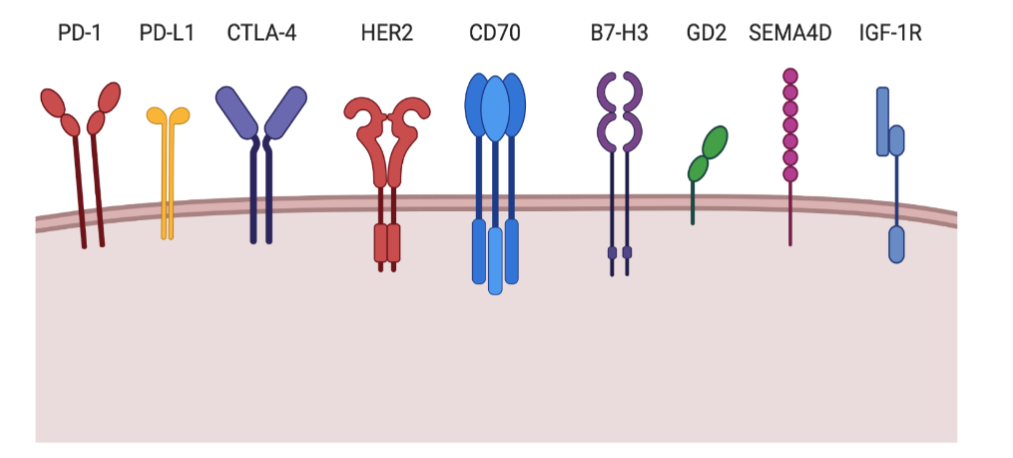
Figure 4. Promising immunotherapy targets of interest for osteosarcoma focused CAR-therapy.
Graphical schematic showing surface proteins that show promise for future therapeutic targeting for OSA.
Targets shown:
PD-1, PD-L1, CTLA-4, HER2, CD70, B7-H3, GD2, SEMA4D, IGF-1R
Authors’ contributions:
GMR researched the topic and wrote the paper. YYV assisted in some of the paper writing and edited the paper. EPR and BSM supervised the research and edited the paper. All authors read and approved the final manuscript.
Ethics approval and consent to participate:
Not applicable
Consent for publication:
Not applicable
Availability of data and materials:
Not applicable
Competing interests:
The authors declare that they have no competing interests.
Funding Acknowledgement:
G.M.R. acknowledges funding from NIH grant F30OD030021. B.S.M. acknowledges funding from NIH grants R01AI146009, R01AI161017, P01CA254849, P50CA136393, U24OD026641, U54CA232561, P30CA077598, U54CA268069, Children’s Cancer Research Fund, the Fanconi Anemia Research Fund, and the Randy Shaver Cancer and Community Fund.
Acknowledgements:
Not applicable
References
1. The lectures of Boyer upon diseases of the bones – Digital Collections – National Library of Medicine. https://collections.nlm.nih.gov/catalog/nlm:nlmuid-2544005R-bk.
2. Manchanda, A. S., Narang, R. S. & Mahajan, S. Osteosarcoma: A case report and evaluation. J. Oral Maxillofac. Pathol. 25, 374–375 (2021).
3. Abarrategi, A. et al. Osteosarcoma: Cells-of-Origin, Cancer Stem Cells, and Targeted Therapies. Stem Cells Int. 2016, 3631764 (2016).
4. Ottaviani, G. & Jaffe, N. The etiology of osteosarcoma. Cancer Treat. Res. 152, 15–32 (2009).
5. Mirabello, L., Troisi, R. J. & Savage, S. A. Osteosarcoma incidence and survival rates from 1973 to 2004: data from the Surveillance, Epidemiology, and End Results Program. Cancer 115, 1531–1543 (2009).
6. Ottaviani, G. & Jaffe, N. The epidemiology of osteosarcoma. Cancer Treat. Res. 152, 3–13 (2009).
7. Biermann, J. S. et al. Bone cancer. J. Natl. Compr. Canc. Netw. 11, 688–723 (2013).
8. Lindsey, B. A., Markel, J. E. & Kleinerman, E. S. Osteosarcoma Overview. Rheumatol Ther 4, 25–43 (2017).
9. Xu, Q., Gao, T., Zhang, B., Zeng, J. & Dai, M. Primary osteosarcoma in elderly patients: A report of three cases. Oncol. Lett. 18, 990–996 (2019).
10. Beird, H. C. et al. Osteosarcoma. Nat Rev Dis Primers 8, 77 (2022).
11. Wadhwa, N. Osteosarcoma: Diagnostic dilemmas in histopathology and prognostic factors. Indian J. Orthop. 48, 247–254 (2014).
12. Bentzen, S. M. et al. Prognostic factors in osteosarcomas. A regression analysis. Cancer 62, 194–202 (1988).
13. Misaghi, A., Goldin, A., Awad, M. & Kulidjian, A. A. Osteosarcoma: a comprehensive review. SICOT J 4, 12 (2018).
14. Tiwari, A. Current concepts in surgical treatment of osteosarcoma. J Clin Orthop Trauma 3, 4–9 (2012).
15. Smeland, S. et al. Scandinavian Sarcoma Group Osteosarcoma Study SSG VIII: prognostic factors for outcome and the role of replacement salvage chemotherapy for poor histological responders. Eur. J. Cancer 39, 488–494 (2003).
16. Petrilli, A. S. et al. Increased survival, limb preservation, and prognostic factors for osteosarcoma. Cancer 68, 733–737 (1991).
17. Jeffree, G. M., Price, C. H. & Sissons, H. A. The metastatic patterns of osteosarcoma. Br. J. Cancer 32, 87–107 (1975).
18. Serpico, R. et al. Metastasis of osteosarcoma to the abdomen: A report of two cases and a review of the literature. Case Rep. Oncol. 14, 647–658 (2021).
19. Sheng, G., Gao, Y., Yang, Y. & Wu, H. Osteosarcoma and metastasis. Front. Oncol. 11, 780264 (2021).
20. Mohseny, A. B. et al. Functional characterization of osteosarcoma cell lines provides representative models to study the human disease. Lab. Invest. 91, 1195–1205 (2011).
21. Kapałczyńska, M. et al. 2D and 3D cell cultures – a comparison of different types of cancer cell cultures. Arch. Med. Sci. 14, 910–919 (2018).
22. Ye, L., Swingen, C. & Zhang, J. Induced pluripotent stem cells and their potential for basic and clinical sciences. Curr. Cardiol. Rev. 9, 63–72 (2013).
23. Takahashi, K. & Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 (2006).
24. Pang, L. K., Pena, M., Zhao, R. & Lee, D.-F. Modeling of osteosarcoma with induced pluripotent stem cells. Stem Cell Res. 49, 102006 (2020).
25. Lee, D.-F. et al. Modeling familial cancer with induced pluripotent stem cells. Cell 161, 240–254 (2015).
26. Zhou, R. et al. Modeling Osteosarcoma Using Li-Fraumeni Syndrome Patient-derived Induced Pluripotent Stem Cells. J. Vis. Exp. (2018) doi:10.3791/57664.
27. Tu, J. et al. Hereditary retinoblastoma iPSC model reveals aberrant spliceosome function driving bone malignancies. Proc. Natl. Acad. Sci. U. S. A. 119, e2117857119 (2022).
28. Jewell, B. E. et al. Patient-derived iPSCs link elevated mitochondrial respiratory complex I function to osteosarcoma in Rothmund-Thomson syndrome. PLoS Genet. 17, e1009971 (2021).
29. Becklin, K. L. et al. Developing Bottom-Up Induced Pluripotent Stem Cell Derived Solid Tumor Models Using Precision Genome Editing Technologies. CRISPR J 5, 517–535 (2022).
30. Baxter, M. et al. Phenotypic and functional analyses show stem cell-derived hepatocyte-like cells better mimic fetal rather than adult hepatocytes. J. Hepatol. 62, 581–589 (2015).
31. Katt, M. E., Placone, A. L., Wong, A. D., Xu, Z. S. & Searson, P. C. In Vitro Tumor Models: Advantages, Disadvantages, Variables, and Selecting the Right Platform. Front Bioeng Biotechnol 4, 12 (2016).
32. Friedrich, J., Seidel, C., Ebner, R. & Kunz-Schughart, L. A. Spheroid-based drug screen: considerations and practical approach. Nat. Protoc. 4, 309–324 (2009).
33. LaBarbera, D. V., Reid, B. G. & Yoo, B. H. The multicellular tumor spheroid model for high-throughput cancer drug discovery. Expert Opin. Drug Discov. 7, 819–830 (2012).
34. Arai, K., Sakamoto, R., Kubota, D. & Kondo, T. Proteomic approach toward molecular backgrounds of drug resistance of osteosarcoma cells in spheroid culture system. Proteomics 13, 2351–2360 (2013).
35. Gebhard, C., Gabriel, C. & Walter, I. Morphological and Immunohistochemical Characterization of Canine Osteosarcoma Spheroid Cell Cultures. Anat. Histol. Embryol. 45, 219–230 (2016).
36. Kundu, B. et al. Mechanical Property of Hydrogels and the Presence of Adipose Stem Cells in Tumor Stroma Affect Spheroid Formation in the 3D Osteosarcoma Model. ACS Appl. Mater. Interfaces 11, 14548–14559 (2019).
37. Gebhard, C. et al. Comparative proteome analysis of monolayer and spheroid culture of canine osteosarcoma cells. J. Proteomics 177, 124–136 (2018).
38. Freeman, F. E., Burdis, R., Mahon, O. R., Kelly, D. J. & Artzi, N. A Spheroid Model of Early and Late-Stage Osteosarcoma Mimicking the Divergent Relationship between Tumor Elimination and Bone Regeneration. Adv. Healthc. Mater. 11, e2101296 (2022).
39. Rimann, M. et al. An in vitro osteosarcoma 3D microtissue model for drug development. J. Biotechnol. 189, 129–135 (2014).
40. Fischetti, T., Di Pompo, G., Baldini, N., Avnet, S. & Graziani, G. 3D Printing and Bioprinting to Model Bone Cancer: The Role of Materials and Nanoscale Cues in Directing Cell Behavior. Cancers 13, (2021).
41. Safhi, A. Y. Three-Dimensional (3D) Printing in Cancer Therapy and Diagnostics: Current Status and Future Perspectives. Pharmaceuticals 15, (2022).
42. Contessi Negrini, N. et al. An Osteosarcoma Model by 3D Printed Polyurethane Scaffold and In Vitro Generated Bone Extracellular Matrix. Cancers 14, (2022).
43. Delgrosso, E. et al. 3D bioprinted osteosarcoma model for experimental boron neutron capture therapy (BNCT) applications: Preliminary assessment. J. Biomed. Mater. Res. B Appl. Biomater. 111, 1571–1580 (2023).
44. Ma, Y. et al. The Dual Effect of 3D-Printed Biological Scaffolds Composed of Diverse Biomaterials in the Treatment of Bone Tumors. Int. J. Nanomedicine 18, 293–305 (2023).
45. Kersten, K., de Visser, K. E., van Miltenburg, M. H. & Jonkers, J. Genetically engineered mouse models in oncology research and cancer medicine. EMBO Mol. Med. 9, 137–153 (2017).
46. Poste, G. & Fidler, I. J. The pathogenesis of cancer metastasis. Nature 283, 139–146 (1980).
47. Asai, T. et al. Establishment and characterization of a murine osteosarcoma cell line (LM8) with high metastatic potential to the lung. Int. J. Cancer 76, 418–422 (1998).
48. Khanna, C. et al. An orthotopic model of murine osteosarcoma with clonally related variants differing in pulmonary metastatic potential. Clin. Exp. Metastasis 18, 261–271 (2000).
49. Luu, H. H. et al. An orthotopic model of human osteosarcoma growth and spontaneous pulmonary metastasis. Clin. Exp. Metastasis 22, 319–329 (2005).
50. McAllister, R. M. et al. Cultivation in vitro of cells derived from a human osteosarcoma. Cancer 27, 397–402 (1971).
51. Manara, M. C. et al. Reversal of malignant phenotype in human osteosarcoma cells transduced with the alkaline phosphatase gene. Bone 26, 215–220 (2000).
52. Du, L., Fan, Q., Tu, B., Yan, W. & Tang, T. Establishment and characterization of a new highly metastatic human osteosarcoma cell line derived from Saos2. Int. J. Clin. Exp. Pathol. 7, 2871–2882 (2014).
53. Billiau, A. et al. Human interferon: mass production in a newly established cell line, MG-63. Antimicrob. Agents Chemother. 12, 11–15 (1977).
54. Wulf-Goldenberg, A., Hoffmann, J., Becker, M., Brzezicha, B. & Walther, W. Patient-Derived Xenografts from Solid Tumors (PDX) for Models of Metastasis. Methods Mol. Biol. 2294, 43–58 (2021).
55. Szewczyk, M., Lechowski, R. & Zabielska, K. What do we know about canine osteosarcoma treatment? Review. Vet. Res. Commun. 39, 61–67 (2015).
56. Simpson, S. et al. Comparative review of human and canine osteosarcoma: morphology, epidemiology, prognosis, treatment and genetics. Acta Vet. Scand. 59, 71 (2017).
57. Bulaklak, K. & Gersbach, C. A. The once and future gene therapy. Nat. Commun. 11, 5820 (2020).
58. Broadhead, M. L., Clark, J. C. M., Choong, P. F. M. & Dass, C. R. Making gene therapy for osteosarcoma a reality. Expert Rev. Anticancer Ther. 10, 477–480 (2010).
59. Aubrey, B. J., Strasser, A. & Kelly, G. L. Tumor-suppressor functions of the TP53 pathway. Cold Spring Harb. Perspect. Med. 6, (2016).
60. Mirabello, L. et al. Germline TP53 variants and susceptibility to osteosarcoma. J. Natl. Cancer Inst. 107, (2015).
61. Phelan, A., Elliott, G. & O’Hare, P. Intercellular delivery of functional p53 by the herpesvirus protein VP22. Nat. Biotechnol. 16, 440–443 (1998).
62. Densmore, C. L. et al. Growth suppression of established human osteosarcoma lung metastases in mice by aerosol gene therapy with PEI-p53 complexes. Cancer Gene Ther. 8, 619–627 (2001).
63. Ganjavi, H. et al. Adenovirus-mediated p53 gene therapy in osteosarcoma cell lines: sensitization to cisplatin and doxorubicin. Cancer Gene Ther. 13, 415–419 (2006).
64. Tsuchiya, H., Mori, Y., Ueda, Y., Okada, G. & Tomita, K. Sensitization and caffeine potentiation of cisplatin cytotoxicity resulting from introduction of wild-type p53 gene in human osteosarcoma. Anticancer Res. 20, 235–242 (2000).
65. Song, S. U. & Boyce, F. M. Combination treatment for osteosarcoma with baculoviral vector mediated gene therapy (p53) and chemotherapy (adriamycin). Exp. Mol. Med. 33, 46–53 (2001).
66. Ye, S. et al. p53 overexpression increases chemosensitivity in multidrug-resistant osteosarcoma cell lines. Cancer Chemother. Pharmacol. 77, 349–356 (2016).
67. Ballatori, S. E. & Hinds, P. W. Osteosarcoma: prognosis plateau warrants retinoblastoma pathway targeted therapy. Signal Transduct. Target. Ther. 1, 16001 (2016).
68. Engeland, K. Cell cycle regulation: p53-p21-RB signaling. Cell Death Differ. 29, 946–960 (2022).
69. Lee, E. Y. et al. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature 359, 288–294 (1992).
70. Zhang, W., Moore, L. & Ji, P. Mouse models for cancer research. Chin. J. Cancer 30, 149–152 (2011).
71. Williams, B. O. et al. Cooperative tumorigenic effects of germline mutations in Rb and p53. Nat. Genet. 7, 480–484 (1994).
72. Feugeas, O. et al. Loss of heterozygosity of the RB gene is a poor prognostic factor in patients with osteosarcoma. J. Clin. Oncol. 14, 467–472 (1996).
73. Ren, W. & Gu, G. Prognostic implications of RB1 tumour suppressor gene alterations in the clinical outcome of human osteosarcoma: a meta-analysis. Eur. J. Cancer Care 26, (2017).
74. Xu, H. J. et al. Enhanced tumor suppressor gene therapy via replication-deficient adenovirus vectors expressing an N-terminal truncated retinoblastoma protein. Cancer Res. 56, 2245–2249 (1996).
75. Craig, C. et al. Effects of adenovirus-mediated p16INK4A expression on cell cycle arrest are determined by endogenous p16 and Rb status in human cancer cells. Oncogene 16, 265–272 (1998).
76. Carmeliet, P. VEGF as a key mediator of angiogenesis in cancer. Oncology 69 Suppl 3, 4–10 (2005).
77. Mei, J. et al. VEGF-siRNA silencing induces apoptosis, inhibits proliferation and suppresses vasculogenic mimicry in osteosarcoma in vitro. Exp. Oncol. 30, 29–34 (2008).
78. Peng, N. et al. Silencing of VEGF inhibits human osteosarcoma angiogenesis and promotes cell apoptosis via VEGF/PI3K/AKT signaling pathway. Am. J. Transl. Res. 8, 1005–1015 (2016).
79. Liu, T.-C. et al. APE1 distinguishes DNA substrates in exonucleolytic cleavage by induced space-filling. Nat. Commun. 12, 601 (2021).
80. Wang, D., Luo, M. & Kelley, M. R. Human apurinic endonuclease 1 (APE1) expression and prognostic significance in osteosarcoma: enhanced sensitivity of osteosarcoma to DNA damaging agents using silencing RNA APE1 expression inhibition. Mol. Cancer Ther. 3, 679–686 (2004).
81. Dai, N. et al. Alteration of the microRNA expression profile in human osteosarcoma cells transfected with APE1 siRNA. Neoplasma 60, 384–394 (2013).
82. Liang, W., Li, C., Li, M., Wang, D. & Zhong, Z. MicroRNA-765 sensitizes osteosarcoma cells to cisplatin via downregulating APE1 expression. Onco. Targets. Ther. 12, 7203–7214 (2019).
83. Wang, D., Zhong, Z.-Y., Li, M.-X., Xiang, D.-B. & Li, Z.-P. Vector-based Ape1 small interfering RNA enhances the sensitivity of human osteosarcoma cells to endostatin in vivo. Cancer Sci. 98, 1993–2001 (2007).
84. Behjati, S. et al. Recurrent mutation of IGF signalling genes and distinct patterns of genomic rearrangement in osteosarcoma. Nat. Commun. 8, 15936 (2017).
85. Farzaei, M. H., Bahramsoltani, R., Rahimi, R., Abbasabadi, F. & Abdollahi, M. A Systematic Review of Plant-Derived Natural Compounds for Anxiety Disorders. Curr. Top. Med. Chem. 16, 1924–1942 (2016).
86. Papachristou, D. J., Batistatou, A., Sykiotis, G. P., Varakis, I. & Papavassiliou, A. G. Activation of the JNK-AP-1 signal transduction pathway is associated with pathogenesis and progression of human osteosarcomas. Bone 32, 364–371 (2003).
87. Dass, C. R., Khachigian, L. M. & Choong, P. F. M. c-Jun Is critical for the progression of osteosarcoma: proof in an orthotopic spontaneously metastasizing model. Mol. Cancer Res. 6, 1289–1292 (2008).
88. Dass, C. R., Khachigian, L. M. & Choong, P. F. M. c-Jun knockdown sensitizes osteosarcoma to doxorubicin. Mol. Cancer Ther. 7, 1909–1912 (2008).
89. Tang, Q. et al. miR-223/Hsp70/JNK/JUN/m iR-223 feedback loop modulates the chemoresistance of osteosarcoma to cisplatin. Biochem. Biophys. Res. Commun. 497, 827–834 (2018).
90. Ren, L. & Khanna, C. Role of ezrin in osteosarcoma metastasis. Adv. Exp. Med. Biol. 804, 181–201 (2014).
91. Ren, L. et al. Dysregulation of ezrin phosphorylation prevents metastasis and alters cellular metabolism in osteosarcoma. Cancer Res. 72, 1001–1012 (2012).
92. Khanna, C. et al. The membrane-cytoskeleton linker ezrin is necessary for osteosarcoma metastasis. Nat. Med. 10, 182–186 (2004).
93. Lo Vasco, V. R., Leopizzi, M., Puggioni, C. & Della Rocca, C. Ezrin silencing remodulates the expression of Phosphoinositide-specific Phospholipase C enzymes in human osteosarcoma cell lines. J. Cell Commun. Signal. 8, 219–229 (2014).
94. Wu, M.-S. et al. CDC20 and its downstream genes: potential prognosis factors of osteosarcoma. Int. J. Clin. Oncol. 24, 1479–1489 (2019).
95. Wang, H., Liu, Z., Wu, P., Wang, H. & Ren, W. NUSAP1 Accelerates Osteosarcoma Cell Proliferation and Cell Cycle Progression via Upregulating CDC20 and Cyclin A2. Onco. Targets. Ther. 14, 3443–3454 (2021).
96. Liu Y., Ding J.-Y., Shen W.-L., Zhao X. & Fan S.-W. [Knockdown of cyclin A2 expression by small interfering RNA in MG-63 cells]. Zhonghua Zhong Liu Za Zhi 29, 670–675 (2007).
97. Shekhar, R. et al. The microRNAs miR-449a and miR-424 suppress osteosarcoma by targeting cyclin A2 expression. J. Biol. Chem. 294, 4381–4400 (2019).
98. Mahmood, N., Mihalcioiu, C. & Rabbani, S. A. Multifaceted Role of the Urokinase-Type Plasminogen Activator (uPA) and Its Receptor (uPAR): Diagnostic, Prognostic, and Therapeutic Applications. Front. Oncol. 8, 24 (2018).
99. Haeckel, C. et al. Protease expression in dedifferentiated parosteal osteosarcoma. Arch. Pathol. Lab. Med. 123, 213–221 (1999).
100. Matsuyama, A., Wood, G. A., Speare, R., Schott, C. R. & Mutsaers, A. J. Prognostic significance of the urokinase plasminogen activator system in tissue and serum of dogs with appendicular osteosarcoma. PLoS One 17, e0273811 (2022).
101. Dass, C. R. et al. Downregulation of uPAR confirms link in growth and metastasis of osteosarcoma. Clin. Exp. Metastasis 22, 643–652 (2005).
102. Borgatti, A. et al. Safe and Effective Sarcoma Therapy through Bispecific Targeting of EGFR and uPAR. Mol. Cancer Ther. 16, 956–965 (2017).
103. de Bock, C. E. et al. Inhibition of urokinase receptor gene expression and cell invasion by anti-uPAR DNAzymes in osteosarcoma cells. FEBS J. 272, 3572–3582 (2005).
104. Endo-Munoz, L. et al. Progression of Osteosarcoma from a Non-Metastatic to a Metastatic Phenotype Is Causally Associated with Activation of an Autocrine and Paracrine uPA Axis. PLoS One 10, e0133592 (2015).
105. Jia, S.-F. et al. Eradication of osteosarcoma lung metastases following intranasal interleukin-12 gene therapy using a nonviral polyethylenimine vector. Cancer Gene Ther. 9, 260–266 (2002).
106. Worth, L. L., Jia, S. F., Zhou, Z., Chen, L. & Kleinerman, E. S. Intranasal therapy with an adenoviral vector containing the murine interleukin-12 gene eradicates osteosarcoma lung metastases. Clin. Cancer Res. 6, 3713–3718 (2000).
107. Tsuji, H. et al. Adenovirus-mediated in vivo B7-1 gene transfer induces anti-tumor immunity against pre-established primary tumor and pulmonary metastasis of rat osteosarcoma. Cancer Gene Ther. 9, 747–755 (2002).
108. Charissoux, J. L., Grossin, L., Leboutet, M. J. & Rigaud, M. Treatment of experimental osteosarcoma tumors in rat by herpes simplex thymidine kinase gene transfer and ganciclovir. Anticancer Res. 19, 77–80 (1999).
109. Ramnaraine, M. et al. Direct and bystander killing of sarcomas by novel cytosine deaminase fusion gene. Cancer Res. 63, 6847–6854 (2003).
110. Ternovoi, V. V., Curiel, D. T., Smith, B. F. & Siegal, G. P. Adenovirus-mediated p53 tumor suppressor gene therapy of osteosarcoma. Lab. Invest. 86, 748–766 (2006).
111. Li, H. et al. FOXP1 drives osteosarcoma development by repressing P21 and RB transcription downstream of P53. Oncogene 40, 2785–2802 (2021).
112. Martínez-Reyes, I. & Chandel, N. S. Cancer metabolism: looking forward. Nat. Rev. Cancer 21, 669–680 (2021).
113. Berger, M. F. & Mardis, E. R. The emerging clinical relevance of genomics in cancer medicine. Nat. Rev. Clin. Oncol. 15, 353–365 (2018).
114. Li, Q. & Kang, C. Mechanisms of Action for Small Molecules Revealed by Structural Biology in Drug Discovery. Int. J. Mol. Sci. 21, (2020).
115. Liu, G.-H., Chen, T., Zhang, X., Ma, X.-L. & Shi, H.-S. Small molecule inhibitors targeting the cancers. MedComm (2020) 3, e181 (2022).
116. Cohen, P., Cross, D. & Jänne, P. A. Kinase drug discovery 20 years after imatinib: progress and future directions. Nat. Rev. Drug Discov. 20, 551–569 (2021).
117. Steeghs, N., Nortier, J. W. R. & Gelderblom, H. Small molecule tyrosine kinase inhibitors in the treatment of solid tumors: an update of recent developments. Ann. Surg. Oncol. 14, 942–953 (2007).
118. Li, S. Anlotinib: A Novel Targeted Drug for Bone and Soft Tissue Sarcoma. Front. Oncol. 11, 664853 (2021).
119. Wang, G. et al. Anlotinib, a novel small molecular tyrosine kinase inhibitor, suppresses growth and metastasis via dual blockade of VEGFR2 and MET in osteosarcoma. Int. J. Cancer 145, 979–993 (2019).
120. Wang, G. et al. Anlotinib Reverses Multidrug Resistance (MDR) in Osteosarcoma by Inhibiting P-Glycoprotein (PGP1) Function In Vitro and In Vivo. Front. Pharmacol. 12, 798837 (2021).
121. Long, Z.-Y. et al. Effective treatment of anlotinib in giant delayed pulmonary metastasis of osteosarcoma: a case report and literature review. Ann. Palliat. Med. 10, 7073–7082 (2021).
122. Tian, S. et al. YN968D1 is a novel and selective inhibitor of vascular endothelial growth factor receptor-2 tyrosine kinase with potent activity in vitro and in vivo. Cancer Sci. 102, 1374–1380 (2011).
123. Aoyama, T. & Yoshikawa, T. Targeted therapy: Apatinib – new third-line option for refractory gastric or GEJ cancer. Nat. Rev. Clin. Oncol. 13, 268–270 (2016).
124. Liu, K. et al. Apatinib promotes autophagy and apoptosis through VEGFR2/STAT3/BCL-2 signaling in osteosarcoma. Cell Death Dis. 8, e3015 (2017).
125. Xie, L. et al. Apatinib for Advanced Osteosarcoma after Failure of Standard Multimodal Therapy: An Open Label Phase II Clinical Trial. Oncologist 24, e542–e550 (2019).
126. Geller, J. I. et al. A study of axitinib, a VEGF receptor tyrosine kinase inhibitor, in children and adolescents with recurrent or refractory solid tumors: A Children’s Oncology Group phase 1 and pilot consortium trial (ADVL1315). Cancer 124, 4548–4555 (2018).
127. Keating, G. M. Axitinib: a review in advanced renal cell carcinoma. Drugs 75, 1903–1913 (2015).
128. Choueiri, T. K. Axitinib, a novel anti-angiogenic drug with promising activity in various solid tumors. Curr. Opin. Investig. Drugs 9, 658–671 (2008).
129. Ranieri, G. et al. Tyrosine-Kinase Inhibitors Therapies with Mainly Anti-Angiogenic Activity in Advanced Renal Cell Carcinoma: Value of PET/CT in Response Evaluation. Int. J. Mol. Sci. 18, (2017).
130. Fioramonti, M. et al. Cabozantinib Affects Osteosarcoma Growth Through A Direct Effect On Tumor Cells and Modifications In Bone Microenvironment. Sci. Rep. 8, 4177 (2018).
131. Wang, J.-H., Zeng, Z., Sun, J., Chen, Y. & Gao, X. A novel small-molecule antagonist enhances the sensitivity of osteosarcoma to cabozantinib in vitro and in vivo by targeting DNMT-1 correlated with disease severity in human patients. Pharmacol. Res. 173, 105869 (2021).
132. Italiano, A. et al. Cabozantinib in patients with advanced Ewing sarcoma or osteosarcoma (CABONE): a multicentre, single-arm, phase 2 trial. Lancet Oncol. 21, 446–455 (2020).
133. Wedge, S. R. et al. AZD2171: a highly potent, orally bioavailable, vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor for the treatment of cancer. Cancer Res. 65, 4389–4400 (2005).
134. Morton, C. L. et al. Combination testing of cediranib (AZD2171) against childhood cancer models by the pediatric preclinical testing program. Pediatr. Blood Cancer 58, 566–571 (2012).
135. Fox, E. et al. A phase 1 trial and pharmacokinetic study of cediranib, an orally bioavailable pan-vascular endothelial growth factor receptor inhibitor, in children and adolescents with refractory solid tumors. J. Clin. Oncol. 28, 5174–5181 (2010).
136. van Cruijsen, H. et al. Phase I evaluation of cediranib, a selective VEGFR signalling inhibitor, in combination with gefitinib in patients with advanced tumours. Eur. J. Cancer 46, 901–911 (2010).
137. Choi, K.-M. et al. Activity-Based Protein Profiling Reveals Potential Dasatinib Targets in Gastric Cancer. Int. J. Mol. Sci. 21, (2020).
138. Marley, K., Gullaba, J., Seguin, B., Gelberg, H. B. & Helfand, S. C. Dasatinib Modulates Invasive and Migratory Properties of Canine Osteosarcoma and has Therapeutic Potential in Affected Dogs. Transl. Oncol. 8, 231–238 (2015).
139. Ding, X. et al. Efficacy and safety of fruquintinib as third- or further-line therapy for patients with advanced bone and soft tissue sarcoma: a multicenter retrospective study. Anticancer Drugs 34, 877–882 (2023).
140. Zhao, Y. & Adjei, A. A. Targeting Angiogenesis in Cancer Therapy: Moving Beyond Vascular Endothelial Growth Factor. Oncologist 20, 660–673 (2015).
141. Iqbal, N. & Iqbal, N. Imatinib: a breakthrough of targeted therapy in cancer. Chemother. Res. Pract. 2014, 357027 (2014).
142. Gai, Q.-J. et al. EPHA2 mediates PDGFA activity and functions together with PDGFRA as prognostic marker and therapeutic target in glioblastoma. Signal Transduct Target Ther 7, 33 (2022).
143. Benini, S. et al. Redundancy of autocrine loops in human osteosarcoma cells. Int. J. Cancer 80, 581–588 (1999).
144. Bond, M. et al. A phase II study of imatinib mesylate in children with refractory or relapsed solid tumors: a Children’s Oncology Group study. Pediatr. Blood Cancer 50, 254–258 (2008).
145. Gobin, B. et al. Imatinib mesylate exerts anti-proliferative effects on osteosarcoma cells and inhibits the tumour growth in immunocompetent murine models. PLoS One 9, e90795 (2014).
146. Suyama, K. & Iwase, H. Lenvatinib: A Promising Molecular Targeted Agent for Multiple Cancers. Cancer Control 25, 1073274818789361 (2018).
147. Gaspar, N. et al. Phase I/II study of single-agent lenvatinib in children and adolescents with refractory or relapsed solid malignancies and young adults with osteosarcoma (ITCC-050)☆. ESMO Open 6, 100250 (2021).
148. Gaspar, N. et al. Lenvatinib with etoposide plus ifosfamide in patients with refractory or relapsed osteosarcoma (ITCC-050): a multicentre, open-label, multicohort, phase 1/2 study. Lancet Oncol. 22, 1312–1321 (2021).
149. Yoon, H.-Y., Park, S., Kim, D. S. & Song, J. W. Efficacy and safety of nintedanib in advanced idiopathic pulmonary fibrosis. Respir. Res. 19, 203 (2018).
150. Zhang, W. et al. Adaptive Fibrogenic Reprogramming of Osteosarcoma Stem Cells Promotes Metastatic Growth. Cell Rep. 24, 1266–1277.e5 (2018).
151. Kallus, S. et al. Nanoformulations of anticancer FGFR inhibitors with improved therapeutic index. Nanomedicine 14, 2632–2643 (2018).
152. Sloan, B. & Scheinfeld, N. S. Pazopanib, a VEGF receptor tyrosine kinase inhibitor for cancer therapy. Curr. Opin. Investig. Drugs 9, 1324–1335 (2008).
153. Kumar, S. et al. Metronomic oral topotecan with pazopanib is an active antiangiogenic regimen in mouse models of aggressive pediatric solid tumor. Clin. Cancer Res. 17, 5656–5667 (2011).
154. Longhi, A. et al. Pazopanib in relapsed osteosarcoma patients: report on 15 cases. Acta Oncol. 58, 124–128 (2019).
155. Wilhelm, S. M. et al. Regorafenib (BAY 73-4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int. J. Cancer 129, 245–255 (2011).
156. Pan, P.-J., Liu, Y.-C. & Hsu, F.-T. Protein Kinase B and Extracellular Signal-Regulated Kinase Inactivation is Associated with Regorafenib-Induced Inhibition of Osteosarcoma Progression In Vitro and In Vivo. J. Clin. Med. Res. 8, (2019).
157. Duffaud, F. et al. Efficacy and safety of regorafenib in adult patients with metastatic osteosarcoma: a non-comparative, randomised, double-blind, placebo-controlled, phase 2 study. Lancet Oncol. 20, 120–133 (2019).
158. Davis, L. E. et al. Randomized Double-Blind Phase II Study of Regorafenib in Patients With Metastatic Osteosarcoma. J. Clin. Oncol. 37, 1424–1431 (2019).
159. Coventon, J. A review of the mechanism of action and clinical applications of sorafenib in advanced osteosarcoma. J Bone Oncol 8, 4–7 (2017).
160. Grignani, G. et al. Sorafenib and everolimus for patients with unresectable high-grade osteosarcoma progressing after standard treatment: a non-randomised phase 2 clinical trial. Lancet Oncol. 16, 98–107 (2015).
161. Papaetis, G. S. & Syrigos, K. N. Sunitinib: a multitargeted receptor tyrosine kinase inhibitor in the era of molecular cancer therapies. BioDrugs 23, 377–389 (2009).
162. Motzer, R. J., Escudier, B., Gannon, A. & Figlin, R. A. Sunitinib: Ten Years of Successful Clinical Use and Study in Advanced Renal Cell Carcinoma. Oncologist 22, 41–52 (2017).
163. Kumar, R. M. R., Arlt, M. J., Kuzmanov, A., Born, W. & Fuchs, B. Sunitinib malate (SU-11248) reduces tumour burden and lung metastasis in an intratibial human xenograft osteosarcoma mouse model. Am. J. Cancer Res. 5, 2156–2168 (2015).
164. Duan, X. L., Guo, J. P., Li, F., Xiu, C. & Wang, H. Sunitinib inhibits PD-L1 expression in osteosarcoma by targeting STAT3 and remodels the immune system in tumor-bearing mice. Future Oncol. 16, 1815–1824 (2020).
165. Penel-Page, M. et al. Off-label use of targeted therapies in osteosarcomas: data from the French registry OUTC’S (Observatoire de l’Utilisation des Thérapies Ciblées dans les Sarcomes). BMC Cancer 15, 854 (2015).
166. McCarthy, E. F. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop. J. 26, 154–158 (2006).
167. Esfahani, K. et al. A review of cancer immunotherapy: from the past, to the present, to the future. Curr. Oncol. 27, S87–S97 (2020).
168. Weiner, L. M., Surana, R. & Wang, S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat. Rev. Immunol. 10, 317–327 (2010).
169. Nelson, P. N. et al. Monoclonal antibodies. Mol. Pathol. 53, 111–117 (2000).
170. Zahavi, D. & Weiner, L. Monoclonal Antibodies in Cancer Therapy. Antibodies (Basel) 9, (2020).
171. Jain, R. K. Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat. Med. 7, 987–989 (2001).
172. Raouf, S., Bertelli, G., Ograbek, A., Field, P. & Tran, I. Real-world use of bevacizumab in metastatic colorectal, metastatic breast, advanced ovarian and cervical cancer: a systematic literature review. Future Oncol. 15, 543–561 (2019).
173. Navid, F. et al. A phase II trial evaluating the feasibility of adding bevacizumab to standard osteosarcoma therapy. Int. J. Cancer 141, 1469–1477 (2017).
174. Pfister, N. T. et al. Mutant p53 cooperates with the SWI/SNF chromatin remodeling complex to regulate VEGFR2 in breast cancer cells. Genes Dev. 29, 1298–1315 (2015).
175. Lowery, C. D. et al. Anti-VEGFR2 therapy delays growth of preclinical pediatric tumor models and enhances anti-tumor activity of chemotherapy. Oncotarget 10, 5523–5533 (2019).
176. Hashimoto, K., Nishimura, S. & Akagi, M. Characterization of PD-1/PD-L1 Immune Checkpoint Expression in Osteosarcoma. Diagnostics (Basel) 10, (2020).
177. Wen, Y. et al. Immune checkpoints in osteosarcoma: Recent advances and therapeutic potential. Cancer Lett. 547, 215887 (2022).
178. Shimizu, T. et al. The effect of immune checkpoint inhibitors on lung metastases of osteosarcoma. J. Pediatr. Surg. 52, 2047–2050 (2017).
179. Dhupkar, P., Gordon, N., Stewart, J. & Kleinerman, E. S. Anti-PD-1 therapy redirects macrophages from an M2 to an M1 phenotype inducing regression of OS lung metastases. Cancer Med. 7, 2654–2664 (2018).
180. Zhang, M.-L., Chen, L., Li, Y.-J. & Kong, D.-L. PD L1/PD 1 axis serves an important role in natural killer cell induced cytotoxicity in osteosarcoma. Oncol. Rep. 42, 2049–2056 (2019).
181. Lussier, D. M. et al. Enhanced T-cell immunity to osteosarcoma through antibody blockade of PD-1/PD-L1 interactions. J. Immunother. 38, 96–106 (2015).
182. Liu, X. et al. Blocking the PD-1/PD-L1 axis enhanced cisplatin chemotherapy in osteosarcoma in vitro and in vivo. Environ. Health Prev. Med. 24, 79 (2019).
183. Yoshida, K. et al. Anti-PD-1 antibody decreases tumour-infiltrating regulatory T cells. BMC Cancer 20, 25 (2020).
184. Tawbi, H. A. et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): a multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 18, 1493–1501 (2017).
185. Le Cesne, A. et al. Programmed cell death 1 (PD-1) targeting in patients with advanced osteosarcomas: results from the PEMBROSARC study. Eur. J. Cancer 119, 151–157 (2019).
186. Geoerger, B. et al. Atezolizumab for children and young adults with previously treated solid tumours, non-Hodgkin lymphoma, and Hodgkin lymphoma (iMATRIX): a multicentre phase 1-2 study. Lancet Oncol. 21, 134–144 (2020).
187. Davis, K. L. et al. Nivolumab in children and young adults with relapsed or refractory solid tumours or lymphoma (ADVL1412): a multicentre, open-label, single-arm, phase 1-2 trial. Lancet Oncol. 21, 541–550 (2020).
188. Boye, K. et al. Pembrolizumab in advanced osteosarcoma: results of a single-arm, open-label, phase 2 trial. Cancer Immunol. Immunother. 70, 2617–2624 (2021).
189. Contardi, E. et al. CTLA-4 is constitutively expressed on tumor cells and can trigger apoptosis upon ligand interaction. Int. J. Cancer 117, 538–550 (2005).
190. Kawano, M., Itonaga, I., Iwasaki, T. & Tsumura, H. Enhancement of antitumor immunity by combining anti-cytotoxic T lymphocyte antigen-4 antibodies and cryotreated tumor lysate-pulsed dendritic cells in murine osteosarcoma. Oncol. Rep. 29, 1001–1006 (2013).
191. Eroglu, Z. et al. Long term survival with cytotoxic T lymphocyte-associated antigen 4 blockade using tremelimumab. Eur. J. Cancer 51, 2689–2697 (2015).
192. Merchant, M. S. et al. Phase I Clinical Trial of Ipilimumab in Pediatric Patients with Advanced Solid Tumors. Clin. Cancer Res. 22, 1364–1370 (2016).
193. Cui, J. et al. Expression and clinical implications of leucine-rich repeat containing 15 (LRRC15) in osteosarcoma. J. Orthop. Res. 38, 2362–2372 (2020).
194. Slemmons, K. K., Mukherjee, S., Meltzer, P., Purcell, J. W. & Helman, L. J. LRRC15 antibody-drug conjugates show promise as osteosarcoma therapeutics in preclinical studies. Pediatr. Blood Cancer 68, e28771 (2021).
195. Hingorani, P. et al. ABBV-085, Antibody-Drug Conjugate Targeting LRRC15, Is Effective in Osteosarcoma: A Report by the Pediatric Preclinical Testing Consortium. Mol. Cancer Ther. 20, 535–540 (2021).
196. Demetri, G. D. et al. First-in-Human Phase I Study of ABBV-085, an Antibody-Drug Conjugate Targeting LRRC15, in Sarcomas and Other Advanced Solid Tumors. Clin. Cancer Res. 27, 3556–3566 (2021).
197. Tabak, S. A., Khalifa, S. E. & Fathy, Y. HER-2 Immunohistochemical Expression in Bone Sarcomas: A New Hope for Osteosarcoma Patients. Open Access Maced J Med Sci 6, 1555–1560 (2018).
198. Park, J. A. & Cheung, N.-K. V. GD2 or HER2 targeting T cell engaging bispecific antibodies to treat osteosarcoma. J. Hematol. Oncol. 13, 172 (2020).
199. Ebb, D. et al. Phase II trial of trastuzumab in combination with cytotoxic chemotherapy for treatment of metastatic osteosarcoma with human epidermal growth factor receptor 2 overexpression: a report from the children’s oncology group. J. Clin. Oncol. 30, 2545–2551 (2012).
200. Wen, Y. H. et al. Epidermal growth factor receptor in osteosarcoma: expression and mutational analysis. Hum. Pathol. 38, 1184–1191 (2007).
201. Sevelda, F. et al. EGFR is not a major driver for osteosarcoma cell growth in vitro but contributes to starvation and chemotherapy resistance. J. Exp. Clin. Cancer Res. 34, 134 (2015).
202. Wang, S. et al. Epidermal growth factor receptor promotes tumor progression and contributes to gemcitabine resistance in osteosarcoma. Acta Biochim. Biophys. Sin. 53, 317–324 (2021).
203. Pahl, J. H. W. et al. Anti-EGFR antibody cetuximab enhances the cytolytic activity of natural killer cells toward osteosarcoma. Clin. Cancer Res. 18, 432–441 (2012).
204. Li, Y.-S., Liu, Q., He, H.-B. & Luo, W. The possible role of insulin-like growth factor-1 in osteosarcoma. Curr. Probl. Cancer 43, 228–235 (2019).
205. Houghton, P. J. et al. Initial testing of a monoclonal antibody (IMC-A12) against IGF-1R by the Pediatric Preclinical Testing Program. Pediatr. Blood Cancer 54, 921–926 (2010).
206. Kolb, E. A. et al. R1507, a fully human monoclonal antibody targeting IGF-1R, is effective alone and in combination with rapamycin in inhibiting growth of osteosarcoma xenografts. Pediatr. Blood Cancer 55, 67–75 (2010).
207. Malempati, S. et al. Phase I/II trial and pharmacokinetic study of cixutumumab in pediatric patients with refractory solid tumors and Ewing sarcoma: a report from the Children’s Oncology Group. J. Clin. Oncol. 30, 256–262 (2012).
208. Kolb, E. A. et al. Initial testing (stage 1) of glembatumumab vedotin (CDX-011) by the pediatric preclinical testing program. Pediatr. Blood Cancer 61, 1816–1821 (2014).
209. Kopp, L. M. et al. Phase II trial of the glycoprotein non-metastatic B-targeted antibody-drug conjugate, glembatumumab vedotin (CDX-011), in recurrent osteosarcoma AOST1521: A report from the Children’s Oncology Group. Eur. J. Cancer 121, 177–183 (2019).
210. Smeester, B. A. et al. SEMA4C is a novel target to limit osteosarcoma growth, progression, and metastasis. Oncogene 39, 1049–1062 (2020).
211. Lussier, D. M., Johnson, J. L., Hingorani, P. & Blattman, J. N. Combination immunotherapy with α-CTLA-4 and α-PD-L1 antibody blockade prevents immune escape and leads to complete control of metastatic osteosarcoma. J Immunother Cancer 3, 21 (2015).
212. Xie, M. et al. Identification of genes contributing to cisplatin resistance in osteosarcoma cells. FEBS Open Bio 13, 164–173 (2023).
213. Clavijo, P. E. et al. Semaphorin4D Inhibition Improves Response to Immune-Checkpoint Blockade via Attenuation of MDSC Recruitment and Function. Cancer Immunol Res 7, 282–291 (2019).
214. Liu, J. et al. Cancer vaccines as promising immuno-therapeutics: platforms and current progress. J. Hematol. Oncol. 15, 28 (2022).
215. Thamm, D. H. et al. Systemic administration of an attenuated, tumor-targeting Salmonella typhimurium to dogs with spontaneous neoplasia: phase I evaluation. Clin. Cancer Res. 11, 4827–4834 (2005).
216. Hashii, Y. et al. WT1 peptide immunotherapy for cancer in children and young adults. Pediatr. Blood Cancer 55, 352–355 (2010).
217. Himoudi, N. et al. Lack of T-cell responses following autologous tumour lysate pulsed dendritic cell vaccination, in patients with relapsed osteosarcoma. Clin. Transl. Oncol. 14, 271–279 (2012).
218. Finocchiaro, L. M. E., Villaverde, M. S., Gil-Cardeza, M. L., Riveros, M. D. & Glikin, G. C. Cytokine-enhanced vaccine and interferon-β plus suicide gene as combined therapy for spontaneous canine sarcomas. Res. Vet. Sci. 91, 230–234 (2011).
219. Gentschev, I. et al. Characterization and evaluation of a new oncolytic vaccinia virus strain LIVP6.1.1 for canine cancer therapy. Bioengineered 4, 84–89 (2013).
220. Cascini, C. et al. Rewiring innate and adaptive immunity with TLR9 agonist to treat osteosarcoma. J. Exp. Clin. Cancer Res. 42, 154 (2023).
221. Mason, N. J. et al. Immunotherapy with a HER2-Targeting Listeria Induces HER2-Specific Immunity and Demonstrates Potential Therapeutic Effects in a Phase I Trial in Canine Osteosarcoma. Clin. Cancer Res. 22, 4380–4390 (2016).
222. Bashor, C. J., Hilton, I. B., Bandukwala, H., Smith, D. M. & Veiseh, O. Engineering the next generation of cell-based therapeutics. Nat. Rev. Drug Discov. 21, 655–675 (2022).
223. Heymann, M.-F., Lézot, F. & Heymann, D. The contribution of immune infiltrates and the local microenvironment in the pathogenesis of osteosarcoma. Cell. Immunol. 343, 103711 (2019).
224. Lu, Y. et al. Novel Immunotherapies for Osteosarcoma. Front. Oncol. 12, 830546 (2022).
225. Fritzsching, B. et al. CD8+/FOXP3+-ratio in osteosarcoma microenvironment separates survivors from non-survivors: a multicenter validated retrospective study. Oncoimmunology 4, e990800 (2015).
226. Sarnaik, A. A., Hwu, P., Mulé, J. J. & Pilon-Thomas, S. Tumor-infiltrating lymphocytes: A new hope. Cancer Cell 42, 1315–1318 (2024).
227. Park, J. A. & Cheung, N.-K. V. Promise and challenges of T cell immunotherapy for osteosarcoma. Int. J. Mol. Sci. 24, (2023).
228. Casanova, J. M. et al. Tumor-Infiltrating Lymphocytes and Cancer Markers in Osteosarcoma: Influence on Patient Survival. Cancers 13, (2021).
229. Zhou, X., Wu, J., Duan, C. & Liu, Y. Retrospective analysis of adoptive TIL therapy plus anti-PD1 therapy in patients with chemotherapy-resistant metastatic osteosarcoma. J. Immunol. Res. 2020, 7890985 (2020).
230. Wang, C., Li, M., Wei, R. & Wu, J. Adoptive transfer of TILs plus anti-PD1 therapy: An alternative combination therapy for treating metastatic osteosarcoma. J Bone Oncol 25, 100332 (2020).
231. Yu, S. & Yao, X. Advances on immunotherapy for osteosarcoma. Mol. Cancer 23, 192 (2024).
232. Rosenberg, S. A. & Restifo, N. P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348, 62–68 (2015).
233. Rohaan, M. W., Wilgenhof, S. & Haanen, J. B. A. G. Adoptive cellular therapies: the current landscape. Virchows Arch. 474, 449–461 (2019).
234. Watanabe, K. et al. Development of a T-cell receptor multimer with high avidity for detecting a naturally presented tumor-associated antigen on osteosarcoma cells. Cancer Sci. 110, 40–51 (2019).
235. Kirtane, K., Elmariah, H., Chung, C. H. & Abate-Daga, D. Adoptive cellular therapy in solid tumor malignancies: review of the literature and challenges ahead. J Immunother Cancer 9, (2021).
236. Rafei, H., Daher, M. & Rezvani, K. Chimeric antigen receptor (CAR) natural killer (NK)-cell therapy: leveraging the power of innate immunity. Br. J. Haematol. 193, 216–230 (2021).
237. Terlikowska, K. M., Dobrzycka, B. & Terlikowski, S. J. Chimeric Antigen Receptor Design and Efficacy in Ovarian Cancer Treatment. Int. J. Mol. Sci. 22, (2021).
238. Kochenderfer, J. N. et al. Construction and preclinical evaluation of an anti-CD19 chimeric antigen receptor. J. Immunother. 32, 689–702 (2009).
239. Ahmed, N. et al. Immunotherapy for osteosarcoma: genetic modification of T cells overcomes low levels of tumor antigen expression. Mol. Ther. 17, 1779–1787 (2009).
240. Rainusso, N. et al. Immunotherapy targeting HER2 with genetically modified T cells eliminates tumor-initiating cells in osteosarcoma. Cancer Gene Ther. 19, 212–217 (2012).
241. Talbot, L. J. et al. A Novel Orthotopic Implantation Technique for Osteosarcoma Produces Spontaneous Metastases and Illustrates Dose-Dependent Efficacy of B7-H3-CAR T Cells. Front. Immunol. 12, 691741 (2021).
242. Zhang, Q. et al. B7-H3 targeted CAR-T cells show highly efficient anti-tumor function against osteosarcoma both in vitro and in vivo. BMC Cancer 22, 1124 (2022).
243. Hidalgo, L. et al. Switchable CAR T cell strategy against osteosarcoma. Cancer Immunol. Immunother. 72, 2623–2633 (2023).
244. Chulanetra, M. et al. GD2 chimeric antigen receptor modified T cells in synergy with sub-toxic level of doxorubicin targeting osteosarcomas. Am. J. Cancer Res. 10, 674–687 (2020).
245. Fernández, L. et al. Memory T Cells Expressing an NKG2D-CAR Efficiently Target Osteosarcoma Cells. Clin. Cancer Res. 23, 5824–5835 (2017).
246. Huang, G. et al. Genetically modified T cells targeting interleukin-11 receptor α-chain kill human osteosarcoma cells and induce the regression of established osteosarcoma lung metastases. Cancer Res. 72, 271–281 (2012).
247. Wang, Y. et al. Anti-CD166/4-1BB chimeric antigen receptor T cell therapy for the treatment of osteosarcoma. J. Exp. Clin. Cancer Res. 38, 168 (2019).
248. Mensali, N. et al. ALPL-1 is a target for chimeric antigen receptor therapy in osteosarcoma. Nat. Commun. 14, 3375 (2023).
249. Zhang, X., Zhang, H., Lan, H., Wu, J. & Xiao, Y. CAR-T cell therapy in multiple myeloma: Current limitations and potential strategies. Front. Immunol. 14, 1101495 (2023).
S
250. Sterner, R. C. & Sterner, R. M. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 11, 69 (2021).
251. Wang, L., Li, W. & Pan, Y. The Eph/Ephrin system in primary bone tumor and bone cancer pain. Aging 15, 7324–7332 (2023).