





Pharmacokinetics of CLX-155 and Metabolites in Mice
Pharmacokinetics of Single-dose CLX-155 and Metabolites in Female Balb/C Mice
John M. York1, Sophie Kang1, Ava Dalton1, Natasha Boyette1, Mahesh Kandula1, Subbu Apparao Suresh
Introduction
The widely used pyrimidine agent, 5-fluorouracil (5-FU), is a cornerstone for treating various cancers. These include colorectal, breast, gastric, and pancreatic cancers¹. As a mainstay of treatment since the 1990s, 5-FU remains one of the main components of chemotherapy combination regimens as an adjuvant therapy for colorectal cancer². With the worsening oncology drug shortages, there is a need for a greater variety of treatments as these drug shortages can be life-threatening for cancer patients³. Moreover, based on the market size growth predictions, the use of antimetabolites in cancer treatment is predicted to grow 69% from 2023 to 2033. An advantage of using 5-FU and capecitabine for gastrointestinal cancers is the possibility of substituting one for the other in case of shortages. Due to its pharmacokinetic (PK) characteristics (e.g., gastrointestinal (GI) absorption, rapid degradation, short half-life), clinicians must administer 5-FU via a prolonged intravenous (IV) infusion⁴. Moreover, 5-FU also has many adverse effects, including diarrhea, neutropenia, and hand-foot syndrome⁵.
Capecitabine is a commonly used oral cytotoxic agent developed to lessen the toxicities associated with IV 5-FU⁶. It is a prodrug of 5-FU that utilizes a different metabolic pathway than 5-FU and has improved bioavailability. It converts to 5’-deoxy-5-fluorocytidine (5’-DFCR) via carboxylesterase enzymes predominantly in the liver. The enzyme cytidine deaminase, primarily present in the liver, plasma, and tumor tissues, then converts 5’-DFCR to 5’-deoxy-5-fluorouridine (5’-DFUR)⁴. Thymidine phosphorylase, an enzyme that has higher concentrations in solid tumors, metabolizes 5’-DFUR to FU⁴. Due to the localization of thymidine phosphorylase to liver and tumor tissues, this 5-FU prodrug results in less systemic toxicity than IV 5-FU⁴. Additionally, its oral administration provides greater convenience for patients by avoiding catheter insertion and the potential need for hospitalization⁷. However, this agent possesses some limitations, including interpatient pharmacokinetic variability related to liver function⁸. Finally, its dosing requires narrow titration and approximately 50% of patients experience severe hand-foot syndrome and severe GI toxicity⁹.
5’-Deoxy-5-fluorocytidine-caprylate conjugate (CLX-155) is a novel 5’-DFCR prodrug under evaluation as an antitumor agent. CLX-155 is a molecular conjugate of acylated 5’-DFCR linked to caprylate. Intestinal wall esterase hydrolyzes CLX-155 to yield 5’-DFCR and caprylic acid, followed by 5’-DFUR and 5-FU. One conversion step exists between capecitabine and CLX-155; however, unlike capecitabine, CLX-155 does not require hepatic metabolism to generate an active metabolite. Instead, CLX-155 undergoes metabolism in the intestinal wall, offering the potential for less interpatient pharmacokinetic variability. The production of caprylic acid upon CLX-155 hydrolysis may also contribute to antitumor activity, which could thus provide CLX-155 with two active moieties¹⁰˒¹¹. In a previous study, CLX-155 demonstrated tumor growth inhibition in colon cancer xenograft nude mice models comparable to capecitabine in a consistent and dose-dependent manner at half the dose¹². CLX-155’s and capecitabine’s similar efficacy profiles in the colon cancer xenograft nude mice model may be related to the former’s unique PK profile.
Therefore, this study addresses the following research questions: 1) what is the single dose PK of CLX-155, and 2) how does it compare to capecitabine? This study’s objective is to examine the PK profile of CLX-155 and capecitabine, along with the PK profiles of their metabolites 5’-DFCR, 5’-DFUR, and 5-FU, to determine whether the similar efficacy profiles of CLX-155 and capecitabine is due to the unique PK profile of the former. This study compared CLX-155 dosed at 250 mg/kg and 500 mg/kg with capecitabine dosed at 500 mg/kg and 1000 mg/kg.
This paper charts the following course to explore the objective. After detailing the study methods, it presents results, discusses them, and suggests future research.
Methods
STUDY DESIGN
This work followed guidance from Ishitsuka¹³ and Onodera et al.¹⁴ for the conduct of single-dose PK studies in animal models. This study utilized a parallel, single-dose design. It engaged four treatment groups, each with 12 mice. Mice received CLX-155 (Group 1: 250 mg/kg; and Group 2: 500 mg/kg) or capecitabine (Group 3: 500 mg/kg; and Group 4: 1000 mg/kg). Investigators administered study treatment via oral gavage (10 mL/kg) after mice had fasted for 4 hours, followed by providing food 2 hours after dosing.
REGULATIONS AND ANIMALS
This study included 48 female Balb/C mice (6 to 8 weeks old; 20 to 25 g body weight) from Vivo Bio Tech (Hyderabad, India). The Institutional Animal Care and Use Committee (IAEC/JDC/2017-120) reviewed and approved procedures involving animal care and use before conduct. Animal care and use adhered to the principles of the Guide for the Care and Use of Laboratory, 8th Edition, 2010 (National Research Council). The facility conducting the experimentation maintains AAALAC (Association for Assessment and Accreditation of Lab Animal Care International) accreditation.
Animals resided in a continuously monitored temperature and humidity-regulated aseptic and access-controlled environment (target ranges: temperature 22 ± 2°C; relative humidity 60 ± 4%; and 60 air changes per hour), with a 12-hour light/dark cycle, and under barrier (quarantine) conditions. Investigators routinely monitored the entire facility to detect any airborne infections. The animals received an autoclaved commercial diet (Nutrilab Rodent Feed, cylindrical-shaped pellets) and had free access to autoclaved water.
STUDY DRUGS
Drug moieties included CLX-155, capecitabine, 5’-DFCR, 5’-DFUR, and 5-FU. The manufacturer Jubilant Chemsys Ltd. (Noida, India) provided CLX-155, whereas standard commercial sources supplied the other active moieties. All chemicals were of analytical grade.
For CLX-155 administration, the formulation consisted of 31.25 mg/mL or 62.5 mg/mL CLX-155 in 32.5% Capryol 90 and 2.5% Polysorbate 80 in water. For capecitabine administration, the formulation consisted of 50 mg/mL or 100 mg/mL in 0.5% HPMC in 40 mM citrate buffer pH 6.
SAMPLE COLLECTION
Blood sample collection occurred at 0.25, 0.5, 1, 2, 4, 8, 10, and 24 hours post-dosing, followed by immediate placement of blood samples on ice prior to centrifugation and plasma storage at -20°C until bio-analysis using liquid chromatography with tandem mass spectrometry (LC-MS/MS). For LC-MS/MS analysis, investigators used API-4000 system with analyst 1.6.1 and Shimadzu Nextra HPLC system with Xterra Phenyl (150 X 3.9 mm, 5 mm, Waters) column (capecitabine, 5’-DFCR and 5’-DFUR) and Kinetix Hile (50 x 2.1 mm, 2.6 mm, Phoenomenex) column (5-FU). For capecitabine, 5’-DFCR and 5’-DFUR, the mobile phases were acetonitrile and 5 mM ammonium acetate in water. For 5-FU, the mobile phases were acetonitrile and 0.2% formic acid in water. The internal standard was warfarin in all LC-MS/MS analyses, which occurred at 70°F. Mass transition (m/z) used are 360 to 244 (capecitabine), 246 to 130 (5’-DFCR), 245 to 129 (5’-DFUR) and 129 to 42.1 (5-FU). The Lower Limit of Quantitation (LLOQ) ranged from 10.9-11.3 ng/ml.
PHARMACOKINETIC ANALYSIS
The investigators focused primarily on the 5-FU and its precursors, 5’-DFCR and 5’-DFUR, as capecitabine and CLX-155 undergo rapid metabolism after administration. They conducted a noncompartmental analysis using WinNonlin Version 7.0 (Certara, Princeton, NJ) to calculate the following parameters: terminal half-life (t₁/₂β) using at least 3 terminal time points; maximum plasma concentration at a drug achieves in a test area of the body (Cmax); area under the plasma concentration-time curve from zero to last measurable time point (AUC₀-t); area under the plasma concentration-time curve from 0 to infinity (AUC₀-∞); and time Cmax occurred (Tmax).
Results
In the female Balb/C mice following single oral gavage administration of CLX-155 (at 250 and 500 mg/kg) and capecitabine (at 500 and 1000 mg/kg), investigators did not observe any clinical signs of toxicity in both the capecitabine and CLX-155 administered groups. In the capecitabine groups, capecitabine was detectable in the plasma samples obtained at 0.25, 0.5 and 1 hr in the 500 mg/kg group and at 0.5, 1 and 2 hr in the 1000 mg/kg group. Capecitabine was not detectable in the plasma samples obtained at other time points. Due to limited data points, investigators did not calculate the PK parameters of capecitabine in either group. In the case of CLX-155 groups, there were no detectable levels of CLX-155 in all the plasma samples obtained (Table 1).
Table 1. Plasma concentration of capecitabine at various time points following single-dose oral administration of capecitabine at 500 mg/kg and 1000 mg/kg.
| Time Point (h) | Capecitabine 500 mg/kg | Capecitabine 1000 mg/kg |
|---|---|---|
| 0.25 | 0.49 ± 0.08 | BLQ |
| 0.5 | 0.27* | 0.28 ± 0.10 |
| 1.0 | 0.03 ± 0.00 | 0.02 ± 0.01 |
| 2.0 | BLQ | 0.02 ± 0.01 |
| 4.0 | BLQ | BLQ |
| 8.0 | BLQ | BLQ |
| 10.0 | BLQ | BLQ |
| 24.0 | BLQ | BLQ |
Values are mean +/- standard deviation.
*Data derived from 2 animals. BLQ: below the level of quantification.
5-FU, 5’-DFCR and 5’-DFUR were detectable in CLX-155 and capecitabine groups and the PK parameters for 5-FU, 5’-DFUR, and 5’-DFCR are summarized in Table 2.
Table 2. PK parameters for 5-FU, 5’-DFUR, and 5’-DFCR.
| Groups | 5-FU t₁/₂ (hr) | 5-FU Cmax (µg/mL) | 5-FU Tmax (hr) | 5-FU AUC₀-t (µg*hr/mL) | 5’-DFUR t₁/₂ (hr) | 5’-DFUR Cmax (µg/mL) | 5’-DFUR Tmax (hr) | 5’-DFUR AUC₀-t (µg*hr/mL) | 5’-DFCR t₁/₂ (hr) | 5’-DFCR Cmax (µg/mL) | 5’-DFCR Tmax (hr) | 5’-DFCR AUC₀-t (µg*hr/mL) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CLX-155 (250 mg/kg) | 3.19 | 0.57 | 0.5 | 1.29 | 4.75 | 7.53 | 2.0 | 40.5 | 3.36 | 14.4 | 2.0 | 73.1 |
| CLX-155 (500 mg/kg) | NRV | 0.91 | 0.5 | 2.26 | 2.59 | 22.5 | 2.0 | 78.5 | 2.31 | 41.2 | 2.0 | 153 |
| Capecitabine (500 mg/kg) | NRV | 0.99 | 1.0 | 1.34 | 5.88 | 40.0 | 0.50 | 63.5 | 5.82 | 137 | 0.25 | 208 |
| Capecitabine (1000 mg/kg) | NRV | 1.51 | 0.5 | 1.94 | 6.27 | 53.9 | 0.50 | 94.4 | 3.97 | 237 | 0.50 | 321 |
NRV: not reportable value.
OVERALL PROFILE
For CLX-155, the systemic exposure (Cmax and AUC₀-t) of 5-FU, 5’-DFUR, and 5’-DFCR demonstrated proportionality to the administered dose. 5’-DFCR and 5’-DFUR showed a delayed Tmax compared to 5-FU. For capecitabine, the systemic exposure (Cmax and AUC₀-t) of 5-FU, 5’-DFCR, and 5’-DFUR was less dose-proportional (Figures 1 and 2). The Tmax was similar across the dose levels studied.
Figure 1. Single-dose CLX-155 (250 and 500 mg/kg) and capecitabine (500 and 1000 mg/kg) AUC₀-t for (A) 5-FU, (B) 5’-DFUR, and (C) 5’-DFCR
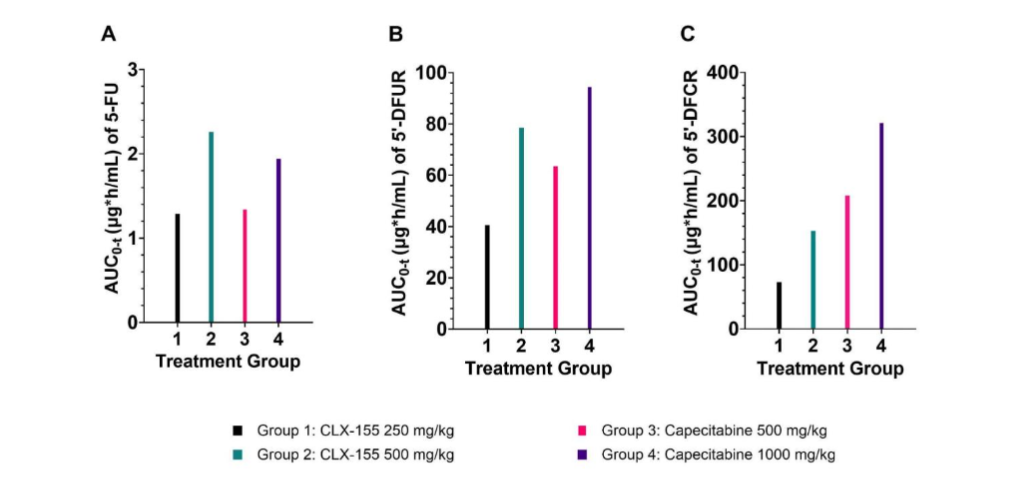
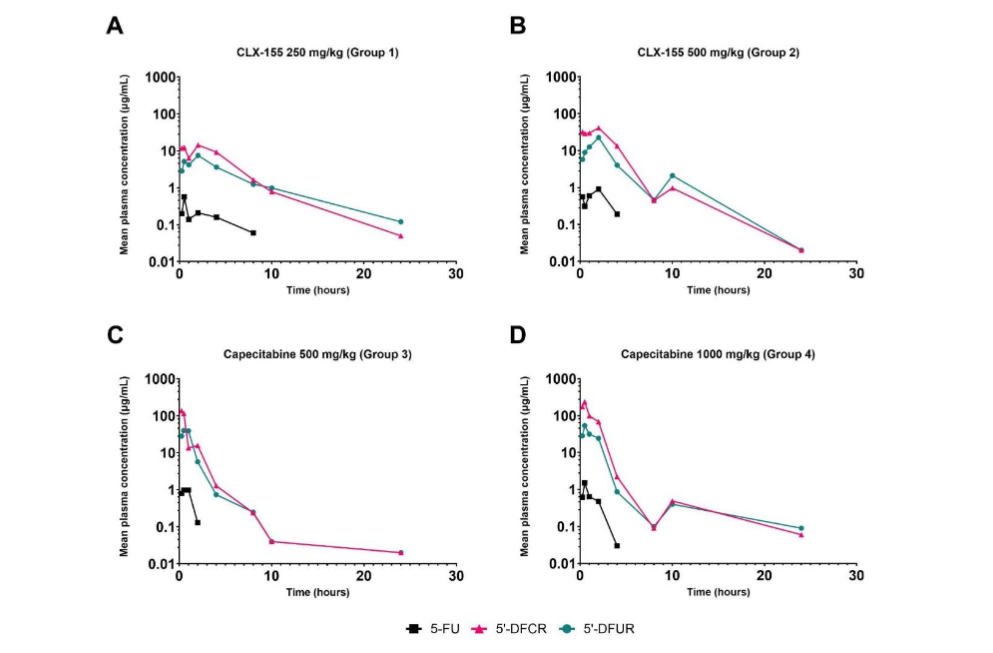
Figure 2. Single-dose mean plasma concentration profiles for CLX-155 at (A) 250 mg/kg and (B) 500 mg/kg and capecitabine at (C) 500 mg/kg and (D) 1000 mg/kg on a logarithmic scale.
CLX-155 demonstrated higher exposure at 500 mg/kg compared to capecitabine at the same dose. CLX-155 and capecitabine appear to display similar plasma Cmax at equivalent dose levels of 500 mg/kg. CLX-155 displayed lower 5’-DFCR and 5’-DFUR Cmax in relation to capecitabine. CLX-155 displayed marginally higher 5’-DFUR and 5-FU plasma AUC₀-t in relation to capecitabine at equivalent doses of 500 mg/kg (Figure 1A and 1B). Investigators found the plasma Cmax of 5-FU to be similar between CLX-155 and capecitabine at the equivalent 500 mg/kg doses.
5-FU
In the CLX-155 groups, administration at 250 mg/kg resulted in a t₁/₂ of 3.19 hours, Cmax of 0.57 µg/mL, Tmax of 0.5 hours, and AUC₀-t of 1.29 µghr/mL for 5-FU as shown in Figure 1A. When investigators increased the dose to 500 mg/kg, the half-life was not reportable, Cmax increased to 0.91 µg/mL, Tmax remained at 0.5 hours, and AUC₀-t increased to 2.26 µghr/mL. Similarly, in the capecitabine groups, 500 mg/kg dosing led to a t₁/₂ that was not reportable, Cmax of 0.99 µg/mL, Tmax of 1.0 hours, and AUC₀-t of 1.34 µghr/mL for 5-FU. At 1000 mg/kg, Cmax increased to 1.51 µg/mL, Tmax remained at 0.5 hours, and AUC₀-t increased to 1.94 µghr/mL.
5’-DFUR
For 5’-DFUR, CLX-155 at 250 mg/kg showed a half-life of 4.75 hours, Cmax of 7.53 µg/mL, Tmax of 2 hours, and AUC₀-t of 40.5 µghr/mL (Figure 1B). For the CLX-155 500 mg/kg group, this metabolite’s t₁/₂ decreased to 2.59 hours, Cmax increased to 22.5 µg/mL, Tmax remained at 2 hours, and AUC₀-t expanded to 78.5 µghr/mL. In the capecitabine groups, 500 mg/kg dosing led to a longer t₁/₂ of 5.88 hours, Cmax of 40 µg/mL, a shorter Tmax of 0.5 hours, and increased AUC₀-t of 63.5 µghr/mL relative to 5’-DFUR. At the 1000 mg/kg dose, the t₁/₂ increased to 6.27 hours, Cmax rose to 53.9 µg/mL, Tmax remained at 0.5 hours, and AUC₀-t expanded to 94.4 µghr/mL for this metabolite.
5’-DFCR
For 5’-DFCR, CLX-155 at 250 mg/kg exhibited a t₁/₂ of 3.36 hours, Cmax of 14.4 µg/mL, Tmax of 2 hours, and AUC₀-t of 73.1 µghr/mL (Figure 1C). Increasing the CLX-155 dose to 500 mg/kg resulted in a lower t₁/₂ of 2.31 hours but a higher Cmax of 41.2 µg/mL, a similar Tmax of 2 hours, and markedly increased AUC₀-t of 153 µghr/mL. In the capecitabine groups, the 500 mg/kg dose showed a longer t₁/₂ of 5.82 hours, higher Cmax of 137 µg/mL, shorter Tmax of 0.25 hours, and increased AUC₀-t of 208 µghr/mL. At 1000 mg/kg of capecitabine, the t₁/₂ decreased to 3.97 hours, Cmax further increased to 237 µg/mL, Tmax remained at 0.5 hours, and AUC₀-t increased substantially to 321 µghr/mL.
Discussion
This study evaluated the single-dose PK of CLX-155 at 250 mg/kg and 500 mg/kg or capecitabine at 500 mg/kg and 1000 mg/kg, and their metabolites 5’-DFCR, 5’-DFUR, and 5-FU in female Balb/C mice, a model widely used in cancer studies¹⁵˒¹⁶. This work’s findings addressed the overarching research questions: 1) what is the single-dose PK of CLX-155, and 2) how does it compare to capecitabine? This research defined the single-dose PK profile (Cmax, Tmax, AUC₀-t, and half-life) for the 5-FU and its precursors at different doses of each of these two prodrugs. These research findings included oral CLX-155’s and capecitabine’s rapid absorption and conversion to 5’-DFCR, 5’-DFUR, and 5-FU. What was unique was that the CLX-155 involved a more sustained-release conversion of the metabolites to 5-FU at half the dose of capecitabine.
Previous studies indicate that patients rapidly absorb capecitabine¹⁷. Orally administered capecitabine enters the portal circulation following intestinal absorption and hydrolysis by the carboxylesterases predominantly in the liver to yield 5’-DFCR¹⁷˒¹⁸. The observation involving capecitabine’s detection in the circulating blood at early time points, in contrast to CLX-155’s non-detection, suggests that CLX-155’s hydrolysis to 5’-DFCR occurred more readily than capecitabine. Since CLX-155 is chemically an ester, the intestinal wall esterase enzymes may hydrolyze this prodrug to yield 5’-DFCR¹². Prior capecitabine work reports high variability in the levels of 5’-DFCR¹⁸. This variability may be due to hepatic blood flow variations and hepatic dysfunction¹⁸. Additionally, the levels and proportions of carboxylesterase isozymes CES1 and CES2 in the liver may affect the efficiency of capecitabine hydrolysis in the liver¹⁹. Capecitabine hydrolysis by CES2 in the intestinal wall drives the GI toxicity profile of capecitabine. Work in male mice has shown diurnal variations in CES1, CES2, cytidine deaminase, and dihydropyrimidine dehydrogenase after capecitabine administration²⁰˒²². This observation may further influence the metabolism of capecitabine and its metabolites²². Previous studies also cite overexpression of cytidine deaminase contributing to capecitabine’s high interpatient variability in pharmacokinetic parameters, which may consequently lead to severe and unexpected toxicities¹⁹.
Other studies indicate that capecitabine’s PK profile reflects this current research’s findings; however, some differences do appear²³. Desmoulin et al. report that after mice received 500 mg/kg of capecitabine, their urine contained 5’-DFCR, 5’-DFUR, and 5-FU²³. These observations indicate the presence of these metabolites in the circulating blood. Based on the measurements in the urine samples, 5’-DFUR appears to be the main metabolite. However, this current study in Balb/c mice demonstrates that 5’-DFCR is capecitabine’s main metabolite. Another study defines the plasma PK profiles of capecitabine, 5’-DFCR, 5’-DFUR, and 5-FU in male C57BL/6J mice after receiving a single, oral dose of capecitabine 500 mg/kg at 4 different time points (ZT1, ZT7, ZT13, and ZT19)²². Akyel et al. find Tmax values for 5-FU, 5’-DFUR, and 5’-DFCR like this study²².
For both CLX-155 and capecitabine, the AUC₀-t and Cmax for all metabolites are dose-proportional. Dose-proportionality is a desirable outcome because it enables investigators to predict the concentration for a given dose and the drug level at various doses. Similar to this study, a previous investigation utilizing a physiologically based PK model in a human cancer xenograft model showed a dose-proportional increase in 5-FU AUC₀-t in blood and tumor tissue after administration of capecitabine²⁴. Previous clinical studies using capecitabine doses of 1250 mg/m² or 1255 mg/m² showed mean Cmax of 5’-DFCR and 5’-DFUR ranging from 2.68 to 5 mg/L and 4.64 to 7.35 mg/L, respectively, which differs from this study where the observation of higher 5’-DFCR Cmax values in comparison to 5’-DFUR²⁵.
One noteworthy observation is that at 500 mg/kg dosing, 5-FU’s AUC₀-t with CLX-155, is 68% greater than that seen with the molar equivalent of capecitabine administration. Similarly, the AUC₀-t of 5’-DFUR with CLX-155 is 24% greater than that with capecitabine (1000 mg/kg). This finding may be due to a more sustained hydrolysis of 5’-DFCR and 5’-DFUR with CLX-155. Such a sustained release of the profile of 5-FU and 5’-DFUR may contribute to the improved efficacy of CLX-155 at lower doses as compared to capecitabine in the human colorectal tumor xenograft nude mice model reported recently¹². Indeed, in clinical observations, prolonged intravenous infusion of 5-FU produced superior response rates compared to bolus schedules²⁶˒²⁷.
Moreover, prior work shows that the 5-FU peak concentration in tumor tissues is 5.5 to 36-fold higher after administration than 5-FU. Such findings imply that giving capecitabine at higher doses than 5-FU can be done safely¹³.
Interestingly, this PK profile might contribute to CLX-155’s antitumor activity as compared with capecitabine. A recent study involving a human colon cancer xenograft model in Foxn1 athymic nude mice compares CLX-155 (125, 250, and 500 mg/kg/day) and capecitabine (1000 mg/kg/day)¹². This work finds that CLX-155 exerts statistically significant, dose-dependent tumor growth inhibition as compared to vehicle control (p<0.0001)¹². It also shows that CLX-155 at 500 mg/kg/day shows similar antitumor activity as capecitabine at 1000 mg/kg/day¹². It also finds that all CLX-155 animals survived, whereas two capecitabine mice experienced toxicity and death¹².
While capecitabine’s indication is for the treatment of colorectal and breast cancers, its adverse event profile, including hand-foot syndrome and gastrointestinal discomfort, limits its use¹. The higher gastrointestinal discomfort seen with capecitabine might be related to its metabolism by the intestinal CES2²⁸.
In contrast, CLX-155 has the potential to produce comparable efficacy at lower doses as compared to capecitabine¹². Besides the extended 5-FU AUC₀-t PK profile with CLX-155, the improved efficacy of CLX-155 in the xenograft model may result from the potential antitumor activity of caprylic acid released by CLX-155 hydrolysis²⁹˒³⁰. Moreover, caprylic acid may also contribute to the extended PK profile of 5-FU derived from CLX-155 due to its emulsification properties that improve bioavailability and drug delivery through the lymphatic system and tumor targeting.
The finding that CLX-155 at 500 mg/kg produces a similar antitumor activity to capecitabine at 1000 mg/kg may lend to a more acceptable adverse effect profile and improve patient adherence and compliance. However, such a supposition will need further animal and human safety studies. Also, when confirmed in clinical studies, such a potentially improved profile of CLX-155 may allow extending the use of CLX-155 to other solid tumors, including pancreatic and prostate tumors. Thus, this current PK study’s findings contribute to a better understanding of the CLX-155’s single-dose PK behavior. Such observations, along with multiple dose studies, can guide development strategies in both the preclinical and clinical settings. Accordingly, studies will help characterize the dose-response relationships, therapeutic efficacy, and safety profiles.
LIMITATIONS
This study does have some limitations, as might be expected with such preclinical work. Because this study involved a single-dose investigation, investigators cannot completely characterize CLX-155’s PK parameters. This investigation was not in a cancer model and, therefore, cannot account for the effects tumors may have on the PK of CLX-155, capecitabine, and their metabolites. Further, investigating multiple doses over time might help define the profile better. Also, variations in human versus mice drug PK may impact the efficacy and toxicity profile.
Next, the mice models may not completely reflect the complexity of colorectal cancers, including differences in types of tumors and heterogeneity. As mice are the subjects of this study, it is difficult to forecast full translatability to humans, dose predictions, and the clinical implications of this data. Mice received doses of CLX-155 and capecitabine via oral gavage, which does not consider the dissolution characteristics of a tablet formation. This work did not take caprylic acid into account when considering metabolites of CLX-155. CLX-155 yields caprylic acid as part of hydrolysis, and this metabolite may contribute to antitumor activity. Accordingly, further work needs to explore caprylic acid’s oncologic effects and how this may impact CLX-155’s oncologic effects.
Finally, this work does not predict safety. Future study considerations include those characterizing the multiple dose PK of CLX-155, capecitabine, and their metabolites, as well as studies characterizing the safety profile of CLX-155.
Conclusion
This single-dose study evaluated the PK profile of CLX-155 in a mouse model with colorectal cancer. CLX-155 demonstrated dose-proportional systemic exposure for 5-FU, 5’-DFUR, and 5’-DFCR. This agent exhibited higher systemic exposure of 5-FU, 5’-DFCR, and 5’-DFUR at a 500 mg/kg dose compared to capecitabine at the same dose. Tmax remained similar across dose levels for both CLX-155 and capecitabine. Capecitabine did not show dose-proportional systemic exposure for 5-FU, 5’-DFUR, and 5’-DFCR; it had less than proportional dose levels compared to the metabolite concentrations. CLX-155 and capecitabine both experience rapid absorption following oral administration and are converted to metabolites 5’-DFCR, 5’-DFUR, and 5-FU.
This single-dose PK study highlights CLX-155’s efficiency in converting to active metabolites and a sustained-release. Such characteristics might contribute to its antitumor activity and potentially lead to safety benefits due to a lower dose needed than capecitabine. Future studies will help refine CLX’s PK, activity, and safety profile in the preclinical and clinical setting.
Conflict of Interests:
Subbu Apparsundaram, PhD and Mahesh Kandula, MS, MBA are Directors in Cellix Therapeutics.
Natasha Boyette, PharmD is a Senior Research Scientist at Merck, Ava Dalton, PharmD and Yearam Tak, PharmD are Johnson and Johnson Fellows, and Sophie Kang, PharmD is a Merck Fellow.
John York, PharmD, MBA is a Consultant to Cellix Therapeutics, COASTAR Therapeutics, Crestec Therapeutics, HRA Rare Disease, JD Biosciences, Reviva Pharmaceuticals and Teikoku Pharma USA.
Funding Statement:
None.
Acknowledgements:
CLX-155 Patent US10858349
References
1. Drug Summary. Accessed Mar 29, 2024. https://www.pdr.net/drug-summary/?drugLabelId=Xeloda-capecitabine-2039
2. Vodenkova S, Buchler T, Cervena K, Veskrnova V, Vodicka P, Vymetalkova V. 5-fluorouracil and other fluoropyrimidines in colorectal cancer: Past, present and future. Pharmacol Ther. 2020;206:107 447. doi:10.1016/j.pharmthera.2019.107447
3. Edgardo S. Santos et al., Drug Shortages in Oncology: ASCO Clinical Guidance for Alternative Treatments. JCO Oncol Pract. 20, 19-32(2024). doi:10.1200/OP.23.00545
4. Walko CM, Lindley C. Capecitabine: A Review. Clin Ther. 2005;27(1). doi:10.1016/j.clinthera
5. Fluorouracil (Systemic). Lexi-Drugs. Hudson, OH: Lexicomp, 2024. http://online.lexi.com/. Updated Mar 27, 2024. Accessed Mar 29, 2024.
6. Jacobs BAW, Deenen MJ, Joerger M, et al. Pharmacokinetics of Capecitabine and Four Metabolites in a Heterogeneous Population of Cancer Patients: A Comprehensive Analysis. CPT Pharmacometrics Syst Pharmacol. 2019;8(12): 940-950. doi:10.1002/psp4.12474.
7. Mineur L, Vazquez L, Belkacemi M, et al. Capecitabine/Mitomycin versus 5-Fluorouracil/Mitomycin in Combination with Simultaneous Integrated Boost Intensity-Modulated Radiation Therapy for Anal Cancer. Curr Oncol. 2023;30(9):8563-8574. doi:10.3390/ curroncol30090621
8. Reigner B, Blesch K, Weidekamm E. Clinical Pharmacokinetics of Capecitabine. Clin Pharmacokinet. 2001;40(2):85-104. doi:10.2165/00 003088-200140020-00002
9. Visacri MB, Duarte NC, Lima T de M, et al. Adverse reactions and adherence to capecitabine: A prospective study in patients with gastrointestinal cancer. J Oncol Pharm Pract. 2022; 28(2):326-336. doi:10.1177/1078155221989420
10. Kobashi N, Matsumoto H, Zhao S, et al. The Thymidine Phosphorylase Imaging Agent 123I-IIMU Predicts the Efficacy of Capecitabine. J Nucl Med. 2016;57(8):1276-1281. doi:10.2967/jnumed .115.165811
11. Jóźwiak M, Filipowska A, Fiorino F, Struga M. Anticancer activities of fatty acids and their heterocyclic derivatives. Eur J Pharmacol. 2020;87 1:172937. doi:10.1016/j.ejphar.2020.172937
12. Boyette N, Dalton A, Tak Y, et al. CLX-155: A Novel, Oral 5-FU Prodrug Displaying Antitumor Activity in Human Colon Cancer Xenograft Model in Nude Mice. Med Res Arch. 2024;12(6). doi:10.1 8103/mra.v12i6.5219
13. Ishitsuka H. Capecitabine: preclinical pharmacology studies. Invest New Drugs. 2000;18 (4):343-354. doi:10.1023/a:1006497231579
14. Onodera H, Kuruma I, Ishitsuka H, Horii I. Pharmacokinetic Study of Capecitabine in Monkeys and Mice; Species Differences in Distribution of the Enzymes Responsible for its Activation to 5-FU. Drug Metab Pharmacokinet. 2000;15(5):439-451. doi:10.2133/dmpk.15.439
15. Potter M. History of the BALB/c family. Curr Top Microbiol Immunol. 1985;122:1-5. doi:10.100 7/978-3-642-70740-7_1
16. Amini A, Safdari Y, Tash Shamsabadi F. Near-Infrared Fluorescence Imaging of EGFR-Overexpressing Tumors in the Mouse Xenograft Model Using scFv-IRDye800CW and Cetuximab-IRDye800CW. Mol Imaging. 2022;2022:9589820. doi:10.1155/2022/9589820
17. Mackean M, Planting A, Twelves C, et al. Phase I and pharmacologic study of intermittent twice-daily oral therapy with capecitabine in patients with advanced and/or metastatic cancer. J Clin Oncol. 1998;16(9):2977-2985. doi:10.1200/JCO.1998.16.9.2977
18. Czejka M, Schueller J, Farkouh A, Gruenberger B, Scheithauer W. Plasma disposition of capecitabine and its metabolites 5’DFCR and 5’DFUR in a standard and dose-intensified monotherapy regimen. Cancer Chemother Pharmacol. 2011;67(3):613-619. doi:10.1007/s002 80-010-1363-4
19. Di L. The Impact of Carboxylesterases in Drug Metabolism and Pharmacokinetics. Curr Drug Metab. 2019;20(2):91-102. doi:10.2174/13892002 19666180821094502
20. Maggo G, Grover SC, Grin A. Capecitabine induced colitis. Pathol Res Pract. 2014;210(9):606-8. doi: 10.1016/j.prp.204.05.005
21. Stathopoulos GP, Koutantos J, Lazaki H, Rigatos SK, Stathopoulos J, Deliconstantinos G. Capecitabine (Xeloda) as monotherapy in advanced breast and colorectal cancer: effectiveness and side-effects. Anticancer Res. 2007;27(3B):1653-1656
22. Akyel YK, Ozturk Civelek D, Ozturk Seyhan N, et al. Diurnal Changes in Capecitabine Clock-Controlled Metabolism Enzymes Are Responsible for Its Pharmacokinetics in Male Mice. J Biol Rhythms. 2023;38(2):171-184. doi:10.1177/074873 04221148779
23. Desmoulin F, Gilard V, Malet-Martino M, Martino R. Metabolism of capecitabine, an oral fluorouracil prodrug: (19)F NMR studies in animal models and human urine. Drug Metab Dispos. 2002;30(11):1221-1229. doi:10.1124/dmd.30.11.1221
24. Tsukamoto Y, Kato Y, Ura M, et al. Investigation of 5-FU disposition after oral administration of capecitabine, a triple-prodrug of 5-FU, using a physiologically based pharmacokinetic model in a human cancer xenograft model: comparison of the simulated 5-FU exposures in the tumour tissue between human and xenograft model. Biopharm Drug Dispos. 2001;22(1):1-14. doi:10.1002/bdd.250
25. Reigner B, Blesch K, Weidekamm E. Clinical pharmacokinetics of capecitabine. Clin Pharmacokinet. 2001;40(2):85-104. doi:10.2165/00 003088-20014 0020-00002
26. Tebbutt NC, Cattell E, Midgley R, Cunningham D, Kerr D. Systemic treatment of colorectal cancer. Eur J Cancer. 2002;38(7):1000-1015. doi:10.1016/s0959-8049(02)00062-x
27. Piedbois P, Rougier P, et al. Meta-analysis Group In Cancer, Efficacy of intravenous continuous infusion of fluorouracil compared with bolus administration in advanced colorectal cancer. J Clin Oncol. 1998;16(1):301-308. doi:10.1200/JCO. 1998.16.1.301
28. Quinney SK, Sanghani SP, Davis WI, et al. Hydrolysis of capecitabine to 5′-deoxy-5-fluorocytidine by human carboxylesterases and inhibition by loperamide. J Pharmacol Exp Ther. 2005;313(3):10 11-1016. doi:10.1124/jpet.104.081265
29. Banerjee S, Kundu A. Lipid-drug conjugates: a potential nanocarrier system for oral drug delivery applications. Daru. 2018;26(1):65-75. doi:10.1007/ s40199-018-0209-1
30. Irby D, Du C, Li F. Lipid-Drug Conjugate for Enhancing Drug Delivery. Mol Pharm. 2017;14(5):1 325-1338. doi:10.1021/acs.molpharmaceut.6b01027