

Advancing Cancer Drug Discovery: Bypassing Synthesis Bottlenecks
Bypassing the Synthesis Bottleneck: A Resource-Stratified Framework for Advancing Cancer Drug Discovery
Dexter Achu Mosoh¹˒²*
- Department of Biomedical Engineering, Indian Institute of Technology Ropar, Rupnagar, Punjab 140001, India
- Professor Wagner A. Vendrame’s Laboratory, Horticultural Sciences Department, University of Florida, Institute of Food and Agricultural Sciences, 2550 Hull Rd., W.M. Fifield Hall, Gainesville, FL 32611, USA
OPEN ACCESS
PUBLISHED: 28 February 2026
CITATION: Mosoh, D.A., 2026. Bypassing the Synthesis Bottleneck: A Resource-Stratified Framework for Advancing Cancer Drug Discovery. Medical Research Archives, [online] 14(2).
COPYRIGHT: © 2026 European Society of Medicine. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
ISSN 2375-1924
Abstract
Despite the proliferation of validated oncogenic targets, the translation of bioactive natural products and high-throughput screening hits into clinical candidates is frequently stalled by the “Synthesis Bottleneck” the prohibitive cost and technical difficulty of optimizing complex chemical scaffolds. This literature-based review addresses this critical impasse by proposing a paradigm shift from rigid structure-replication to function-mimicry, exemplified by the evolution of the complex marine natural product Halichondrin B to the simplified clinical drug Eribulin. It presents a resource-stratified framework that empowers researchers across the funding spectrum to navigate synthetic intractability. For resource-constrained academic laboratories, it highlights the emergence and revolution of ultra-large “make-on-demand” virtual libraries and pharmacophore hopping. For mid-tier biotechnology firms, it explores fragment-based drug discovery and free energy perturbation modeling to de-risk synthesis. For well-resourced pharmaceutical entities, it discusses the closed-loop integration of generative AI with autonomous robotic synthesis. Finally, it examines modality switching specifically Proteolysis-Targeting Chimeras (PROTACs) and covalent inhibitors as a strategic “escape hatch” for targets that remain refractory to traditional small-molecule optimization. By matching specific computational and experimental tools to available resources, this framework aims to democratize the discovery of developable cancer therapies and rescue promising biological hypotheses from the graveyard of intractable chemistry.
Keywords: Synthetic accessibility; Function-oriented synthesis; Oncology drug discovery; Make-on-demand libraries; PROTACs; Generative AI; Lead optimization; Eribulin.
Introduction
A stark dichotomy currently characterizes the discovery of novel cancer therapeutics: while understanding of oncogenic drivers has expanded exponentially, the attrition rate for new chemical entities entering clinical development remains prohibitively high. This inefficiency is often referred to as the Oncology Paradox, in which high attrition rates are driven less by a lack of biological efficacy than by the synthetic intractability of potent lead compounds. Historically, oncology has relied heavily on natural products (NPs) such as taxanes and vinca alkaloids which possess the structural complexity and three-dimensionality required to engage challenging biological targets. However, the same complexity that confers biological potency frequently renders these molecules synthetically intractable, creating a bottleneck that prevents the optimization of promising hits into viable clinical candidates.
As a result, compelling biological hypotheses are often abandoned in the Valley of Death between hit identification and lead optimization, simply because the chemistry required to refine them is too costly or complex to sustain.
This literature-based review addresses the critical impasse at which biological potential intersects with chemical feasibility. A paradigm shift is advanced from Structure Replication the traditional emphasis on the total synthesis of complex natural products to Function Mimicry, which prioritizes preserving the essential pharmacophore within simplified scaffolds. This approach markedly reduces molecular complexity and cost of goods, as exemplified by the successful evolution of the complex marine sponge metabolite halichondrin B into the clinically approved drug eribulin.
To operationalize this shift, a resource-stratified framework is presented as a tiered toolkit that aligns synthetic strategies with the economic and infrastructural constraints of different research environments. For resource-limited academic laboratories, this includes leveraging ultra-large, billion-scale make-on-demand libraries to bypass in-house synthesis. In contrast, well-resourced pharmaceutical organizations may integrate generative artificial intelligence (AI) and autonomous robotic platforms to enable rapid, closed-loop optimization workflows.
For targets where high-affinity small molecules remain synthetically inaccessible despite scaffold simplification, the strategic value of modality switching is also examined. By transitioning to event-driven pharmacological approaches such as proteolysis-targeting chimeras (PROTACs) or covalent inhibitors, simpler and lower-affinity warheads can be employed to achieve therapeutic efficacy, thereby circumventing the need for complex, high-affinity binders. Overall, this review aims to provide actionable strategies for researchers across academic and industrial settings to overcome synthetic bottlenecks and accelerate the translation of chemically intractable leads into effective cancer therapies.
1. The Graveyard of Good Ideas
1.1 THE ONCOLOGY PARADOX
Historically, natural products have served as the cornerstone of cancer chemotherapy. An analysis of approved small-molecule anticancer drugs reveals that approximately 80% are either natural products, derivatives thereof, or synthetic mimetics based on natural pharmacophores. The reliance on these compounds stems from their inherent structural diversity and biological relevance; evolution has selected these molecules to interact with biological macromolecules with high specificity and potency.
Notable examples include the taxanes (e.g., paclitaxel) and vinca alkaloids (e.g., vinblastine), which remain essential components of standard-of-care regimens for malignancies ranging from breast and ovarian cancer to leukemias.
However, this reliance presents a fundamental conflict: the oncology paradox. While evolutionary pressure selects for biological potency, it does not select for synthetic accessibility or drug-like physicochemical properties. Natural products typically possess higher molecular weights, more oxygen atoms, fewer nitrogen atoms, and a significantly higher fraction of sp3-hybridized bridgehead atoms and chiral centers compared to synthetic libraries. For instance, the original isolation of halichondrin B, a polyether macrolide with 32 chiral centers, required massive quantities of marine sponge to yield milligrams of compound, creating a supply crisis that nearly halted its development into the drug eribulin. Consequently, the field faces a dichotomy where the most potent biological starting points are often the most chemically intractable.
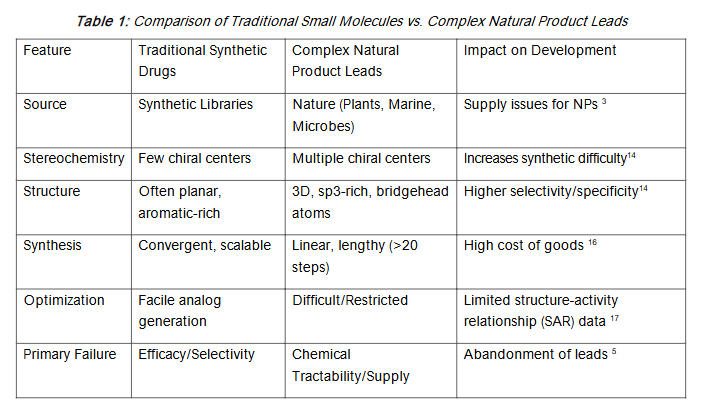
1.2 DEFINING THE HURDLE
The transition from a bioactive hit to a clinical candidate requires optimization of efficacy, pharmacokinetics (PK), and safety a process heavily dependent on chemical tractability. “Intractable” hits are often characterized by structural features that preclude efficient modification, such as multiple stereogenic centers, complex macrocycles, or unstable cores.
The “Intractable” Hit Structural complexity often correlates with synthetic difficulty. The bridgehead double bonds found in complex terpenes (e.g., taxol) represent high ring-strain energy and torsional distortion, posing formidable challenges for chemical synthesis and modification. Furthermore, many natural product scaffolds violate traditional “rule of five” (Ro5) guidelines for oral bioavailability, falling into the “beyond Rule of 5” (bRo5) chemical space, which includes complex modalities like proteolysis-targeting chimeras (PROTACs) and macrocycles. While these complex structures can engage ‘undruggable’ targets such as protein-protein interactions (PPIs) at flat, featureless interfaces, their development is often hindered by lengthy, low-yielding syntheses that limit the rapid generation of structure-activity relationship (SAR) data.
The Metric: Quantifying Difficulty To objectively define “too hard,” computational metrics have been developed to assess synthetic accessibility (SA). Approaches like the Synthetic Accessibility Score (SAS) and SCScore (Synthetic Complexity Score) utilize machine learning and reaction databases to estimate the complexity of synthesizing a target molecule. These metrics move beyond intuitive assessments by medicinal chemists, providing quantitative data on fragment frequency, stereo-complexity, and ring topology.

1.3 THE CONSEQUENCE: THE ATTRITION FUNNEL
The inability to efficiently optimize complex leads contributes significantly to the high attrition rates observed in oncology drug development. Historically, attrition rates in oncology have been reported as high as 82% to 95%, significantly exceeding those in other therapeutic areas.
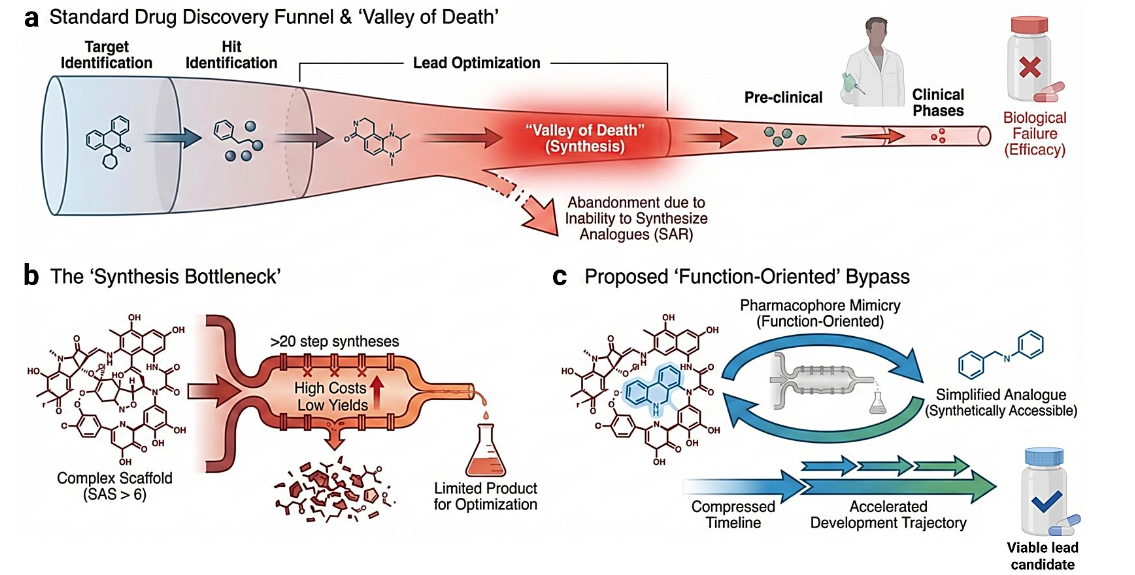
While early failures were often attributed to poor pharmacokinetics, modern attrition is increasingly driven by lack of efficacy and safety issues that emerge in late-stage trials. This shift underscores the consequences of the synthesis bottleneck: when a lead compound is synthetically intractable, medicinal chemists cannot easily modify the scaffold to eliminate toxicophores or improve target selectivity without destroying biological activity. Consequently, promising biological hypotheses are often abandoned, or suboptimal candidates are advanced, leading to failure in Phase II or Phase III trials. This “Valley of Death” in anticancer drug development represents a failure not necessarily of biology, but of the chemical tools available to validate that biology.
1.4 THE RESOURCE DIVIDE
The synthesis bottleneck creates a distinct resource divide in drug discovery. The development of complex bRo5 molecules involves rising costs of goods due to lengthy syntheses, specialized formulations, and stringent storage requirements. While large pharmaceutical entities may possess the resources to navigate these challenges exemplified by the massive investment required to develop the 62-step synthesis of eribulin, academic labs and smaller biotech firms are disproportionately affected.
The “age of feasibility” in total synthesis has demonstrated that while almost any molecule can be made given infinite resources, making them efficiently enough for drug development remains a distinct challenge. This disparity widens the gap between target discovery and therapeutic application. Academic groups often identify novel bioactive natural products or targets but lack the synthetic capacity to optimize them into viable drug candidates, leaving potential therapies for “undruggable” targets stranded in the literature.
2. The Core Philosophy: Structure-Replication vs. Function-Mimicry
2.1 THE “TOTAL SYNTHESIS” TRAP
The traditional approach to developing natural products into therapeutics has often been characterized by an adherence to structure-replication, wherein synthetic chemists endeavor to reproduce the exact chemical architecture of a bioactive hit. This paradigm, historically rooted in the structural elucidation of complex molecules like reserpine and vitamin B12, frequently leads to a “Total Synthesis” trap. While total synthesis remains a powerful vehicle for confirming structural assignments and demonstrating chemical prowess, it often proves inefficient for drug discovery. The intricate scaffolds of natural products evolved for biological defense rather than human therapy can demand lengthy, low-yielding sequences that are impractical for generating the large libraries required for structure-activity relationship (SAR) studies or for scalable manufacturing. For instance, the synthesis of complex polyketides and terpenes, such as spirastrellolide A or ingenol, involves numerous steps to install multiple stereocenters and macrocyclic features, often resulting in bottlenecks that hinder the rapid delivery of clinical candidates. This fixation on precise replication can stifle innovation, leaving potential treatments stranded in academic literature rather than progressing to the clinic.
2.2 THE “FUNCTION-ORIENTED” SHIFT
In response to the limitations of structure-replication, a paradigm shift toward function-oriented synthesis (FOS) has emerged. This philosophy advocates for distilling a complex natural product down to its essential pharmacophoric elements the specific structural features responsible for its biological activity while discarding auxiliary complexity. By focusing on function rather than structure, chemists can design simplified scaffolds that are more chemically accessible, tunable, and drug-like. FOS strategies include the design of “bryologs” that mimic the function of the complex marine natural product bryostatin 1 with significantly reduced structural complexity, and the development of simplified probes for targets like protein kinase C (PKC).
This approach aligns with the concept of “lead-oriented synthesis,” which emphasizes the preparation of diverse, lead-like small molecules with properties suitable for optimization. Strategies such as scaffold hopping, molecular editing, and the use of natural product-derived fragments allow for the exploration of chemical space that retains biological relevance but offers superior synthetic tractability. For example, the transformation of the complex alkaloid quinine into diverse scaffolds via selective chemical editing demonstrates the potential to generate novel, functional libraries from natural starting points.
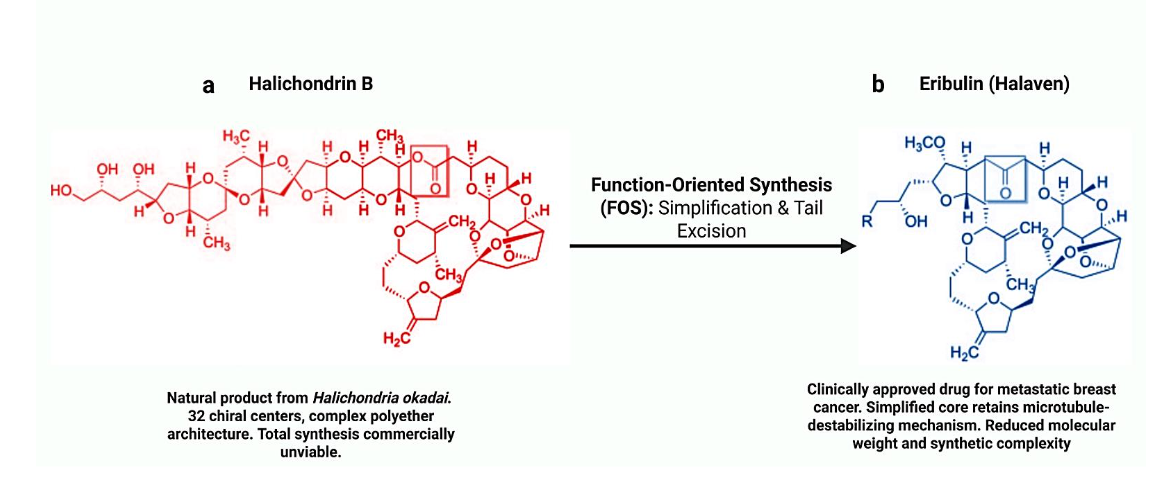
2.3 THE GOLDEN CASE STUDY: HALICHONDRIN B TO ERIBULIN (HALAVEN)
The evolution of Halichondrin B into the clinical drug Eribulin (Halaven) stands as the definitive proof of the FOS philosophy. Halichondrin B, a polyether macrolide isolated from the marine sponge Halichondria okadai, exhibits potent anticancer activity but possesses a formidable structure with 32 chiral centers, making the natural supply impossible and total synthesis daunting.
The Insight: Through extensive SAR studies conducted by Eisai scientists in collaboration with the Kishi group, it was discovered that the biological activity resided primarily in the macrocyclic “right half” of the molecule. The complex polyether “tail,” while structurally impressive, was auxiliary to the tubulin-binding function.
The Result: By discarding the unnecessary tail and focusing on the macrocyclic pharmacophore, scientists developed Eribulin a simplified, fully synthetic analogue that retains the potent microtubule-destabilizing mechanism of the parent natural product. Eribulin (Halaven) was approved by the FDA in 2010 for the treatment of metastatic breast cancer, validating the FOS approach. This transition from an intractable natural product to a manufacturable drug highlights the power of function-mimicry over structure-replication.
3. A Stratified Arsenal: Solutions for Every Resource Level
The financial and infrastructural disparities between academic laboratories, mid-sized biotech firms, and large pharmaceutical companies necessitate a stratified approach to overcoming the synthesis bottleneck. This framework categorizes solutions based on available resources, empowering researchers at every level to access chemical matter for “intractable” targets.
3.1 THE “LOW-TO-NO” BUDGET PORTFOLIO (ACADEMIC LABS / NON-PROFITS)
Philosophy: Do not make; Buy. For academic labs with limited synthetic chemistry infrastructure, the most effective strategy to bypass the synthesis bottleneck is to eliminate synthesis in the early discovery phase. The “Make-on-Demand” revolution has fundamentally altered the landscape of accessible chemical space, allowing researchers to purchase billions of novel, synthesizable compounds directly.
The “Make-on-Demand” Revolution – Traditional virtual screening often relies on “In-Stock” libraries like the ZINC database, which typically contain millions of compounds. While valuable, these collections represent a minute fraction of drug-like chemical space and are biased towards historical medicinal chemistry targets. In contrast, “Make-on-Demand” libraries such as Enamine REAL Space and WuXi GalaXi offer access to tens of billions of compounds.
The Math: These libraries are constructed combinatorially using robust, validated reactions and available building blocks. For example, the Enamine REAL Space contains over 37 billion molecules.
Why it works: Because the synthetic routes are pre-validated (e.g., amide couplings, reductive aminations), the success rate for synthesis is high (>80%), and the cost is low (~$100 per compound) with rapid delivery times (3-4 weeks). This allows academic labs to screen ultra-large libraries virtually and order only the top hits, effectively outsourcing the synthetic risk.
Pharmacophore Hopping – For labs without access to expensive commercial software, open-source tools provide a viable path to finding simpler, purchasable analogues of complex leads. Tools like Pharmit and RDKit allow researchers to define the 3D electrostatic and shape features (pharmacophore) of an intractable hit and screen against public databases to find structurally distinct but functionally similar molecules. This “scaffold hopping” approach can identify novel chemotypes that retain biological activity but possess simpler, more tractable scaffolds.
3.2 THE MID-RANGE BUDGET PORTFOLIO (BIOTECH / FUNDED ACADEMIC)
Philosophy: Grow from simplicity rather than pruning complexity. Biotech companies and well-funded academic groups often have the resources for some medicinal chemistry but must prioritize targets efficiently. The strategy here shifts from finding a perfect replacement to growing a lead from a manageable starting point.
Fragment-Based Drug Discovery (FBDD) – FBDD inverts the traditional problem of “pruning” a complex natural product by starting with a small, chemically simple “fragment” (MW <300 Da) that binds weakly to the target.
Method: These fragments are screened using sensitive biophysical methods (e.g., Nuclear Magnetic Resonance (NMR), Surface Plasmon Resonance (SPR), X-ray crystallography). Once a hit is identified, it is “grown” or “linked” to other fragments to increase affinity while maintaining synthetic tractability.
Cancer Context: A premier example is Venetoclax (ABT-199), a first-in-class anti-apoptotic B-cell lymphoma 2 (Bcl-2) protein inhibitor. The discovery began with fragment screening to identify small molecules binding to the Bcl-2 homology 3 (BH3) domain, a protein-protein interaction site previously considered “undruggable” due to its large surface area. Through structure-guided optimization, these simple fragments were evolved into a potent, clinically approved drug.
DNA-Encoded Library (DEL) – DEL screening is a high-capacity hit-identification platform that enables the simultaneous screening of billions of small molecules in a single tube, providing unprecedented parallel interrogation of chemical space. In DELs, each compound in the library is covalently linked to a unique DNA barcode that serves as a digital surrogate encoding its synthetic history. However, unlike functional assays that directly measure biological activity (e.g., enzyme inhibition), DEL screening primarily reports binding affinity. Consequently, initial hits represent binders rather than confirmed functional leads and therefore require orthogonal follow-up validation.
Method: In a typical DEL screening workflow, the pooled library is incubated with a purified target protein, most commonly immobilized on a solid support. Following incubation, non-binding compounds are removed through stringent washing steps, while high-affinity binders are selectively retained. The DNA barcodes associated with these binders are subsequently amplified by polymerase chain reaction (PCR) and decoded using next-generation sequencing (NGS), enabling rapid identification of the active chemical structures.
Advantage: A key advantage of DEL technology is its capacity to democratize ultra-large-scale screening, allowing mid-sized organizations to access capabilities traditionally limited to large pharmaceutical companies for the identification of novel, synthetically tractable hits suitable for further optimization, while requiring minimal protein consumption and infrastructure investment. Moreover, recent innovations have expanded the scope of DELs beyond traditional formats. These include fragment-based DELs, which integrate principles of fragment-based drug discovery (FBDD) with DEL scale, as well as DNA-encoded dynamic libraries (DEDLs) that leverage target-templated synthesis to uncover novel, drug-like hits, including for targets previously considered undruggable.
Targeted FEP+ (Free Energy Perturbation) – Computational physics has matured to the point where it can reliably predict binding affinity, significantly de-risking synthetic efforts. FEP+ uses molecular dynamics simulations to calculate the free energy difference between ligands binding to a protein.
Application: Instead of synthesizing 100 analogues to find one with improved potency, a biotech can “virtually synthesize” and test them using FEP. This allows for the prioritization of only the top few candidates for physical synthesis, maximizing the return on investment for synthetic chemistry resources.
3.3 THE HIGH-BUDGET PORTFOLIO (PHARMA / MAJOR INSTITUTES)
Philosophy: Closed-loop autonomy. Large pharmaceutical companies and major research institutes can leverage high-end infrastructure to completely automate the “Design-Make-Test-Analyze” (DMTA) cycle, collapsing the timeline from hit to lead.
Generative AI & De Novo Design – Advanced AI models, such as REINVENT, NVIDIA BioNeMo, and graph-based genetic algorithms, are now capable of de novo molecular design.
Constraint-Based Generation: Crucially, these models can be constrained to propose only molecules that are synthetically accessible. By integrating reaction rules and available building blocks into the generative process, the AI avoids proposing “imaginary” molecules that cannot be made.
Optimization: These tools can simultaneously optimize for multiple parameters, such as potency, selectivity, and drug-likeness, effectively serving as a virtual medicinal chemist.
The “Digital-to-Physical” Loop – The ultimate expression of this tier is the integration of AI design with robotic synthesis platforms. Workflow: Generative AI algorithms propose novel ligands, while automated retrosynthesis software predicts viable synthetic pathways; these designs can then be transitioned to robotic platforms including liquid handlers and automated flow reactors to execute the synthesis and accelerate the discovery cycle.
Impact: This closed-loop system enables the autonomous optimization of reaction conditions and the rapid production of chemical libraries. By utilizing high-throughput screening, automated platforms can optimize challenging couplings in days rather than months. This acceleration is critical for complex cancer targets where iterative DMTA (Design-Make-Test-Analyze) cycles are the primary bottleneck to clinical development.
4. Bypassing Synthesis via Modality Switching
4.1 THE LOGIC: CHANGING THE RULES OF ENGAGEMENT
When a potent small molecule inhibitor demands a synthetic route of excessive length (e.g., >20 steps) or complexity (e.g., multiple chiral centers, macrocyclization), the bottleneck often lies in the stringent structural requirements for high-affinity binding. Traditional occupancy-driven inhibitors must maintain prolonged binding to a specific active site to exert a therapeutic effect, necessitating precise complementary interactions that drive synthetic complexity.
The strategic pivot here is to shift from occupancy-driven pharmacology to event-driven or covalent modalities. This shift relaxes the requirement for high-affinity, reversible binding, allowing the use of simpler, more synthetically accessible chemical matter as “warheads” or “handles” rather than highly optimized inhibitors.
4.2 PROTACs & DEGRADERS: EFFICACY FROM SIMPLICITY
The Hack: Proteolysis-Targeting Chimeras (PROTACs) and molecular glues represent a paradigm shift. Unlike inhibitors, PROTACs do not need to bind tightly to the target’s active site to function; they merely need to recruit the target protein to an E3 ubiquitin ligase for subsequent degradation.
Counter-Intuitive Advantage: While PROTACs are typically large, beyond-Rule-of-5 (bRo5) molecules, their event-driven pharmacology allows for the use of ‘sloppy’ or weak-binding warheads. This enables the conversion of simple, low-affinity fragments which would otherwise be ineffective as traditional inhibitors into potent therapeutic ‘handles’ that drive highly efficient, catalytic protein degradation.
Synthetic Implication: The shift toward event-driven pharmacology significantly relaxes the requirement for high-affinity warheads. Instead of optimizing a lead through 10-15 steps to achieve nanomolar potency, a 3-5 step analogue with micromolar affinity can be linked to an E3 ligase ligand to create a potent degrader. However, this shifts the synthetic focus toward linker optimization, where the geometry of the ternary complex rather than simple binding affinity dictates therapeutic success.
Cancer Context: This strategy has been pivotal in targeting ‘undruggable’ proteins such as STAT3 and BRD4, where traditional inhibitors often fail due to a lack of binding pockets or compensatory protein accumulation. For example, bavdegalutamide (ARV-110), an androgen receptor degrader, targets clinically relevant mutants (e.g., T878A, H875Y) that are resistant to standard antiandrogens; by physically eliminating the receptor, it overcomes resistance mechanisms driven by both mutational escape and target overexpression.
4.3 COVALENT INHIBITORS: LOCKING ONTO THE TARGET
Targeted Covalent Inhibition (TCI) provides a strategic alternative to traditional equilibrium-based drug design, allowing for the bypass of complex, high-affinity scaffolds. By forming an irreversible bond with a specific nucleophilic residue most commonly Cysteine, though increasingly Lysine or Tyrosine on the target protein, a covalent inhibitor achieves a duration of action that is decoupled from systemic pharmacokinetics.
Method: Inhibition occurs via a two-step mechanism: first, the molecule forms a reversible, non-covalent complex with the target; second, the electrophilic “warhead” (e.g., acrylamide) is positioned to undergo a bond-forming reaction with the protein.
The Advantage: Since the final bond is irreversible, the initial binding affinity can be significantly lower than that of a reversible drug. This enables the use of simpler, less optimized scaffolds that are “locked” in place by the reactive warhead, resulting in “infinite” residence time that is limited only by the rate of de novo protein synthesis (turnover).
Case Study: KRAS G12C. Long considered ‘undruggable’ due to its lack of deep binding pockets and picomolar affinity for GTP, the KRAS G12C breakthrough relied on a covalent tethering strategy rather than traditional high-affinity competition. Sotorasib (AMG 510) utilizes an acrylamide warhead to covalently modify the mutant Cysteine-12, trapping the protein in its inactive GDP-bound conformation.
This ‘electrophile-first’ approach demonstrates that targeting site-specific reactivity can overcome the limitations of featureless binding sites.
Selectivity & Safety: A critical consideration for covalent inhibitors is minimizing off-target reactivity. To ensure safety, modern targeted covalent inhibitors (TCIs) are optimized so that the warhead reacts only after specific scaffold binding, with activity-based protein profiling (ABPP) serving as a critical tool for verifying proteome-wide selectivity.
5. The Pragmatic Guide: A Resource-Stratified Decision Matrix for Lead Optimization
The transition from a validated hit to a developable lead is the most resource-intensive and failure-prone phase of early drug discovery. A primary cause of attrition at this stage is the synthesis bottleneck, in which chemically complex but biologically promising hits cannot be efficiently optimized. To mitigate this risk and prevent the premature abandonment of viable biological hypotheses, this review proposes a structured, resource-stratified decision matrix that integrates quantitative assessments of synthetic complexity with the availability of computational and experimental resources.
This framework replaces intuition-driven decision-making with a clear, actionable workflow that guides researchers toward the most appropriate optimization strategy for their specific context, ultimately maximizing the likelihood of delivering a synthetically accessible lead candidate.
5.1 PHASE I: QUANTITATIVE ASSESSMENT OF CHEMICAL INTRACTABILITY
The decision to pursue or bypass traditional medicinal chemistry optimization must be data-driven. Upon identifying a validated hit or lead compound, researchers should first quantify its synthetic complexity using established metrics, such as the Synthetic Accessibility Score (SAS) or the SCScore.
A triage threshold is applied at this initial gate. Hits with a SAS 6, or scaffolds predicted to require fewer than ten synthetic steps with reasonable overall yields, may proceed through conventional design-make-test cycles. In contrast, compounds exceeding a SAS of 6, requiring more than ten reaction steps, or exhibiting overall yields below 1% represent a hard stop for standard medicinal chemistry. Historical analyses show that continued optimization of such scaffolds correlates strongly with failure, primarily due to the inability to generate sufficient structure-activity relationship (SAR) data. Compounds exceeding this threshold must therefore exit the linear optimization pipeline and enter alternative workflows.
5.2 PHASE II: PATH SELECTION BASED ON STRUCTURAL DATA AND RESOURCE AVAILABILITY
Once a compound is deemed synthetically intractable, the decision matrix bifurcates based on the availability of structural data and the laboratory’s computational and financial resources.
5.2.1 Ligand-Based Path: Low-Resource Settings
In academic or resource-constrained environments, where high-throughput synthesis and structure determination are often unavailable, the primary objective is to replace the complex lead with a purchasable surrogate. In the absence of a protein-ligand crystal structure, optimization proceeds via a ligand-based strategy.
First, the key steric and electrostatic features of the intractable lead are extracted to define a pharmacophore model using open-source tools such as Pharmit or RDKit. This pharmacophore model is then deployed to screen ultra-large, combinatorial ‘make-on-demand’ spaces such as Enamine REAL or WuXi GalaXi which provide access to billions of molecules with reported synthesis success rates exceeding 80%. This approach effectively converts the internal synthetic burden into a scalable procurement model, enabling laboratories to evaluate hundreds of structurally diverse, simplified analogues at a fraction of the cost of internal development. Consequently, researchers can rapidly re-establish an SAR-driven optimization loop without exhausting specialized synthetic resources on intractable leads.
5.2.2 Structure-Based Path: Mid- to High-Resource Settings
In industrial or well-resourced settings, where protein-ligand crystal structures or high-confidence AlphaFold-derived models are available, optimization follows a structure-based path. This approach utilizes molecular docking integrated with synthetic feasibility filters (e.g., SAS or SCScore) to provide a high-resolution prioritization of analogues, ensuring that only the most thermodynamically favorable and synthetically tractable candidates proceed to advanced simulation or wet-lab validation. For higher-resolution optimization, physics-based methods such as Free Energy Perturbation (FEP+) enable the quantitative affinity ranking of virtual analogues with experimental-grade accuracy. By prioritizing only the top-ranked candidates for physical synthesis, this strategy drastically reduces the number of ‘failed’ analogues produced, ensuring that specialized synthetic resources are reserved for the most promising leads. At the highest resource tier, generative AI models (e.g., REINVENT) are integrated into closed-loop discovery systems. By explicitly constraining the chemical search space to accessible building blocks and validated reaction classes, these models generate candidates that are both potent in silico and directly compatible with automated or semi-automated synthesis platforms, effectively merging molecular design with laboratory execution.
5.3 PHASE III: THE MODALITY ESCAPE HATCH
Should ligand- or structure-based optimization prove insufficient due to featureless binding interfaces or persistent low activity the framework mandates a strategic modality switch. One pathway utilizes targeted protein degradation (PROTACs) to bypass the need for high-affinity occupancy; by leveraging event-driven pharmacology, simple or low-affinity fragments can achieve potent cellular activity through catalytic degradation. Alternatively, for targets possessing accessible nucleophilic residues (e.g., Cysteine), covalent modification can ‘rescue’ reversible hits by maximizing residence time and functional potency, effectively converting a transient interaction into a sustained therapeutic effect.
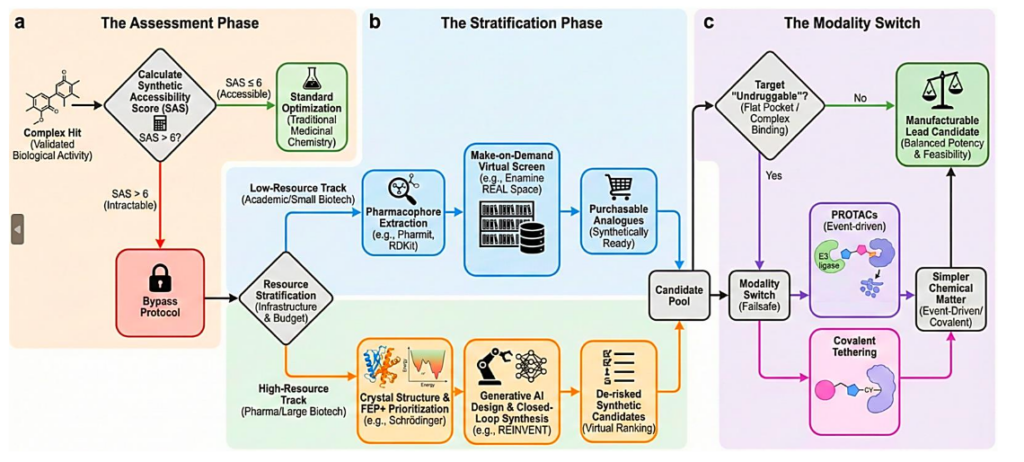
Figure 3 | Workflow for bypassing synthetic intractability during lead optimization.
a, Complexity assessment using Synthetic Accessibility Scores to initiate bypass decisions.
b, Resource-dependent strategies from purchasable libraries to AI-assisted de novo design.
c, Modality switching as a failsafe for targets incompatible with small-molecule optimization.
5.4 OUTCOME: A SYNTHETICALLY ACCESSIBLE LEAD CANDIDATE
By progressing through structured decision gates from quantitative complexity assessment to resource-stratified path selection this matrix provides a high-velocity roadmap for lead optimization. This pragmatic approach de-risks the early discovery pipeline by aligning chemical strategy with operational capacity, thereby minimizing wasted synthetic effort and maximizing the probability that biologically validated hits mature into synthetically tractable, developable lead candidates.
6. Conclusion
The trajectory of oncology drug discovery is being reshaped by a fundamental decoupling of biological function from structural complexity. As demonstrated, the ‘synthesis bottleneck’ is not an immutable barrier, but a strategic inflection point. By pivoting from the rigid imitation of natural architectures to the pragmatic emulation of their pharmacophores, researchers can rescue high-value ‘intractable’ targets previously sidelined by prohibitive synthetic costs. This shift from labor-intensive synthesis to resource-stratified, digital-first optimization ensures that the next generation of cancer therapeutics is defined by clinical impact rather than synthetic difficulty.
The future of drug discovery lies in the synergistic application of diverse modalities that prioritize functional outcome over structural complexity. The transition from occupancy-driven inhibitors to event-driven modalities including PROTACs and molecular glues demonstrates that high-affinity binding is no longer a prerequisite for therapeutic efficacy. This shift enables the use of synthetically accessible chemical matter to achieve potent biological results, effectively bypassing the need for labor-intensive total synthesis. Concurrently, the expanding covalent inhibitor toolbox allows researchers to ‘lock’ simple scaffolds into featureless targets, achieving levels of selectivity and potency that would otherwise require formidable architectural complexity.
A critical evolution in the coming decade will be the democratization of discovery tools, effectively lowering the barrier to entry for complex drug design. The explosive growth of ‘make-on-demand’ combinatorial libraries allows academic and non-profit researchers to access chemical diversity that was previously the exclusive domain of large-scale pharmaceutical enterprises. When integrated with AI-driven retrosynthesis and generative design, these resources minimize the trial-and-error redundancy inherent in traditional lead optimization transforming drug discovery into a more economically sustainable and accessible endeavor.
Looking forward, the successful medicinal chemist will act not merely as a builder of atoms, but as an architect of function. The integration of closed-loop automated synthesis platforms with generative AI will finalize the transition from hypothesis to validation, drastically compressing the hit-to-lead timeline.
Ultimately, the sustainability of oncology R&D depends on elevating ‘developability’ and ‘synthesizability’ to the same level of priority as biological potency. By embracing a stratified, function-first approach, the discovery community can ensure that the next generation of cancer therapeutics is defined not only by their discovery in the lab but by their manufacturability and delivery to the clinic.
Outstanding Questions
- Can AI predict “Functional Tractability” over “Synthetic Accessibility”? Current metrics like SAS quantify the difficulty of making a specific structure. Can next-generation algorithms predict the probability that a complex natural product has a simplified, synthetically accessible analogue that retains biological efficacy?
- Will “Make-on-Demand” libraries cover the chemical space of “Undruggable” targets? While libraries like Enamine REAL Space have expanded to billions of compounds, they are largely dominated by reactions that are easy to automate (e.g., amide couplings). Does this chemical space possess the three-dimensionality and complexity required to disrupt difficult protein-protein interactions in cancer, or does it merely populate the “druggable” genome more densely?
- Is the Eribulin success story replicable via automation? The simplification of Halichondrin B to Eribulin took decades of human insight. Can generative AI and deep learning models accelerate this “pharmacophore distillation” process to months, or does it require a level of biological intuition currently beyond computational reach?
- How will the “democratization” of discovery affect safety profiles? As academic labs and small biotechs gain access to potent screening tools and covalent warheads, will the lack of rigorous, industrial-scale ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) profiling lead to a glut of potent but toxic leads in the literature?
- Can covalent inhibitors target residues beyond Cysteine? Current “escape hatch” strategies rely heavily on cysteine-reactive warheads (e.g., acrylamides). With the emergence of warheads targeting lysine, tyrosine, and aspartate, can we expand the “covalentome” to rescue targets that lack accessible cysteines?
Glossary
- Covalent Inhibitor – A drug molecule equipped with a reactive functional group (warhead) that forms a specific, permanent bond with a target protein residue (often cysteine). This modality allows for high potency and prolonged duration of action even with scaffolds that have lower reversible binding affinity.
- DNA-Encoded Library (DEL) – A technology for the synthesis and screening of combinatorial chemical libraries where each small molecule is covalently attached to a unique DNA barcode. This allows billions of compounds to be mixed and screened against a protein target in a single tube, with hits identified via DNA sequencing.
- Fragment-Based Drug Discovery (FBDD) – A method of finding lead compounds by screening very small molecular fragments (MW <300 Da) that bind weakly to a biological target. Because fragments are simple, they are chemically efficient starting points that can be “grown” or “linked” into potent drugs with optimized properties.
- Free Energy Perturbation (FEP+) – A computational method that uses statistical mechanics and molecular dynamics simulations to predict the binding free energy change between two or more ligands. It allows researchers to “virtually synthesize” and test hundreds of analogues with high accuracy before committing to physical synthesis.
- Function-Oriented Synthesis (FOS) – A synthetic philosophy that aims to recapitulate the biological activity (function) of a complex natural product using simpler scaffolds, rather than replicating the exact chemical structure.
- Make-on-Demand Library – Huge virtual collections of compounds (often billions) that are not physically stored but can be synthesized rapidly (typically 3-4 weeks) upon order. They are constructed computationally using validated building blocks and robust chemical reactions, ensuring a high success rate.
- Pharmacophore – The ensemble of steric and electronic features of a molecule that is necessary to ensure the optimal supramolecular interactions with a specific biological target and to trigger (or block) its biological response.
- Proteolysis-Targeting Chimera (PROTAC) – A heterobifunctional molecule that links a protein of interest (POI) to an E3 ubiquitin ligase. This proximity induces the ubiquitination and subsequent proteasomal degradation of the POI. PROTACs operate via an event-driven mode of action, allowing the degradation of targets that are otherwise difficult to inhibit.
- Synthetic Accessibility Score (SAS) – A computational metric (typically 1 to 10) used to estimate the difficulty of synthesizing a drug-like molecule. It combines analysis of molecular fragments with penalties for structural complexity to prioritize compounds that are feasible to manufacture.
- Molecular glue – A class of small-molecule degraders that induce or stabilize a de novo protein-protein interaction between an E3 ubiquitin ligase and a specific target protein (neosubstrate), thereby promoting ubiquitination and proteasomal degradation.
Data Availability:
All data supporting the findings of this review are included in this article. Further inquiries should be directed to Dexter Achu Mosoh.
Acknowledgments:
This study did not receive any external funding. The corresponding author extends his deepest gratitude to Chief MOSOH Paul Tandong and Chieftess Ateyim Espe MOSOH Ostensia Nkeng of PINYIN (Santa, North-West Region, Cameroon) for their unwavering support, which has been instrumental to his professional growth and this research endeavor. Additionally, the author thanks the reviewer(s) for their thorough review and valuable suggestions that greatly improved this publication. All figures were created with the assistance of BioRender.
Author Contributions:
DM conceptualized this review and provided overall project administration and supervision. DM also developed the methodology, conducted the investigation, performed the formal analysis, curated the data, and created the visualizations. As the principal author, DM was responsible for writing the manuscript, including the original draft and all subsequent revisions and edits.
Competing Interests:
The author declares that there are no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References:
- Kuo Y, Barrett JS. Consideration of the Root Causes in Candidate Attrition During Oncology Drug Development. Clin Pharmacol Drug Dev. 2024; 13(9):952-960.
- Walker I, Newell H. Do molecularly targeted agents in oncology have reduced attrition rates? Nat Rev Drug Discov. 2009;8(1):15.
- Cragg GM, Pezzuto JM. Natural products as a vital source for the discovery of cancer chemotherapeutic and chemopreventive agents. Med Princ Pract. 2016; 25(Suppl. 2):41-59.
- Mann J. Natural products in cancer chemotherapy: past, present and future. Nat Rev Cancer. 2002;2(2):143-148.
- Showell GA, Mills JS. Chemistry challenges in lead optimization: silicon isosteres in drug discovery. Drug Discov Today. 2003;8(12):551-556.
- Wender PA, Verma VA, Paxton TJ, Pillow TH. Function-oriented synthesis, step economy, and drug design. Acc Chem Res. 2008;41(1):40-49.
- Bellmann L, Penner P, Gastreich M, Rarey M. Comparison of Combinatorial Fragment Spaces and Its Application to Ultralarge Make-on-Demand Compound Catalogs. J Chem Inf Model. 2022;62(3):553-566.
- Doveston R, Marsden S, Nelson A. Towards the realisation of lead-oriented synthesis. Drug Discov Today. 2014;19(7):813-819.
- Dai T, Vijayakrishnan S, Szczypiński FT, et al. Autonomous mobile robots for exploratory synthetic chemistry. Nature. 2024;635(8040):890-897.
- Neklesa TK, Winkler JD, Crews CM. Targeted protein degradation by PROTACs. Pharmacol Ther. 2017;174:138-144.
- Mosoh DA. Widely-targeted in silico and in vitro evaluation of veratrum alkaloid analogs as FAK inhibitors and dual targeting of FAK and Hh/SMO pathways for cancer therapy: A critical analysis. Int J Biol Macromol. Published online October 4, 2024:136201.
- Mosoh DA. Recent Advances in Phytochemical Research for Cancer Treatment. IntechOpen; 2024.
- Xiao Z, Morris‐Natschke SL, Lee K. Strategies for the optimization of natural leads to anticancer drugs or drug candidates. Med Res Rev. 2016;36(1):32-91.
- Rodrigues T, Reker D, Schneider P, Schneider G. Counting on natural products for drug design. Nat Chem. 2016;8(6):531-541.
- Huang M, Lu JJ, Ding J. Natural products in cancer therapy: Past, present and future. Nat Prod Bioprospecting. 2021;11(1):5-13.
- Eastgate MD, Schmidt MA, Fandrick KR. On the design of complex drug candidate syntheses in the pharmaceutical industry. Nat Rev Chem. 2017;1(2):0016.
- Truax NJ, Romo D. Bridging the gap between natural product synthesis and drug discovery. Nat Prod Rep. 2020;37(11):1436-1453.
- Liu J, Liu X, Wu J, Li CC. Total synthesis of natural products containing a bridgehead double bond. Chem. 2020;6(3):579-615.
- Yang W, Gadgil P, Krishnamurthy VR, et al. The evolving druggability and developability space: chemically modified new modalities and emerging small molecules. AAPS J. 2020;22(2):21.
- Surade S, Blundell TL. Structural biology and drug discovery of difficult targets: the limits of ligandability. Chem Biol. 2012;19(1):42-50.
- Skoraczyński G, Kitlas M, Miasojedow B, Gambin A. Critical assessment of synthetic accessibility scores in computer-assisted synthesis planning. J Cheminformatics. 2023;15(1):6.
- Zhang Y, Zhang Z, Wen L, Liu Y. Synthetic accessibility scoring and its application to the high-throughput design of energetic molecules. J Mater Chem A. 2025;13(22):16330-16344.
- Yu J, Wang J, Zhao H, et al. Organic compound synthetic accessibility prediction based on the graph attention mechanism. J Chem Inf Model. 2022;62(12):2973-2986.
- Moreno L, Pearson AD. How can attrition rates be reduced in cancer drug discovery? Expert Opin Drug Discov. 2013;8(4):363-368.
- Unibest Industrial. Scaling the Everest of Pharma Synthesis: A Deep Dive into Eribulin’s Journey – Unibest Industrial Co., Ltd. 2024. Accessed February 5, 2026.
- Baran PS. Natural product total synthesis: as exciting as ever and here to stay. J Am Chem Soc. 2018;140(14):4751-4755.
- Tamura T, Kawano M, Hamachi I. Targeted covalent modification strategies for drugging the undruggable targets. Chem Rev. 2025;125(2):1191-1253.
- Chen FE, Huang J. Reserpine: a challenge for total synthesis of natural products. Chem Rev. 2005;105(12):4671-4706.
- Morrill LA, Susick RB, Chari JV, Garg NK. Total Synthesis as a Vehicle for Collaboration. J Am Chem Soc. 2019;141(32):12423-12443.
- Reisman SE, Maimone TJ. Total synthesis of complex natural products: more than a race for molecular summits. Acc Chem Res. 2021;54(8):1815-1816.
- Rishton GM. Natural products as a robust source of new drugs and drug leads: past successes and present day issues. Am J Cardiol. 2008;101(10):S43-S49.
- Thomford NE, Senthebane DA, Rowe A, et al. Natural products for drug discovery in the 21st century: innovations for novel drug discovery. Int J Mol Sci. 2018;19(6):1578.
- Paterson I, Lam NYS. Challenges and discoveries in the total synthesis of complex polyketide natural products. J Antibiot (Tokyo). 2018;71(2):215-233.
- Blakemore DC, Castro L, Churcher I, et al. Organic synthesis provides opportunities to transform drug discovery. Nat Chem. 2018;10(4):383-394.
- Chen K, Wu F, Lei X. Function‐oriented natural product synthesis. Chin J Chem. 2021;39(4):838-854.
- Levin MD, Sarpong R, Wendlandt AE. What is Editing? Acc Chem Res. 2025;58(11):1725-1726.
- Ma C, Lindsley CW, Chang J, Yu B. Rational molecular editing: a new paradigm in drug discovery. J Med Chem. 2024;67(14):11459-11466.
- Morrison KC, Hergenrother PJ. Natural products as starting points for the synthesis of complex and diverse compounds. Nat Prod Rep. 2013;31(1):6-14.
- Melvin JY, Zheng W, Seletsky BM, Littlefield BA, Kishi Y. Case history: discovery of eribulin (HalavenTM), a halichondrin B analogue that prolongs overall survival in patients with metastatic breast cancer. In: Annual Reports in Medicinal Chemistry. Vol 46. Elsevier; 2011:227-241.
- Irwin JJ, Tang KG, Young J, et al. ZINC20 A Free Ultralarge-Scale Chemical Database for Ligand Discovery. J Chem Inf Model. 2020;60(12):6065-6073.
- Lyu J, Irwin JJ, Shoichet BK. Modeling the expansion of virtual screening libraries. Nat Chem Biol. 2023;19(6):712-718.
- Bui L, Djikic-Stojsic T, Bret G, Bihel F, Kellenberger E. Scaffold-Based Libraries Versus Make-on-Demand Space: A Comparative Assessment of Chemical Content. ChemMedChem. 2025;20(24):e202500518.
- Popov KI, Wellnitz J, Maxfield T, Tropsha A. HIt Discovery using docking ENriched by GEnerative Modeling (HIDDEN GEM): A novel computational workflow for accelerated virtual screening of ultra-large chemical libraries. Mol Inform. 2024;43(1):e202300207.
- Kuan J, Radaeva M, Avenido A, Cherkasov A, Gentile F. Keeping pace with the explosive growth of chemical libraries with structure-based virtual screening. WIREs Comput Mol Sci. 2023;13(6):e1678.
- Festel G. Outsourcing chemical synthesis in the drug discovery process. Drug Discov Today. 2011;16(5):237-243.
- Jackson V, Jordan L, Burgin RN, McGaw OJS, Muir CW, Ceban V. Application of Molecular Modeling, Scaffold-Hopping, and Bioisosteric Approaches to the Discovery of New Heterocyclic Picolinamides. J Agric Food Chem. 2022;70(36):11031-11041.
- Muegge I, Bentzien J, Ge Y. Perspectives on current approaches to virtual screening in drug discovery. Expert Opin Drug Discov. 2024;19(10):1173-1183.
- Mishra A, Thakur A, Sharma R, et al. Scaffold hopping approaches for dual-target antitumor drug discovery: opportunities and challenges. Expert Opin Drug Discov. 2024;19(11):1355-1381.
- AlKharboush DF, Kozielski F, Wells G, Porta EOJ. Fragment-based drug discovery: A graphical review. Curr Res Pharmacol Drug Discov. 2025;9:100233.
- Khedkar NR, Sindkhedkar M, Joseph A. Fragment-Based Drug Discovery: Small Fragments, Big Impact Success Stories of Approved Oncology Therapeutics. Bioorganic Chem. 2025;156:108197.
- Bon M, Bilsland A, Bower J, McAulay K. Fragment-based drug discovery the importance of high-quality molecule libraries. Mol Oncol. 2022;16(21):3761-3777.
- Fesik SW. Drugging Challenging Cancer Targets Using Fragment-Based Methods. Chem Rev. 2025;125(6):3586-3594.
- Peterson AA, Liu DR. Small-molecule discovery through DNA-encoded libraries. Nat Rev Drug Discov. 2023;22(9):699-722.
- Song M, Hwang GT. DNA-Encoded Library Screening as Core Platform Technology in Drug Discovery: Its Synthetic Method Development and Applications in DEL Synthesis. J Med Chem. 2020;63(13):6578-6599.
- Poupart J. DNA-Encoded Libraries in Cancer Research: Recent Landmarks and Future Promises. Published online October 16, 2025.
- Gao Y, Liu J, Huang S, Du N, Zhang G, Li Y. Recent advances in DNA-encoded libraries. Chem Commun. 2025;61(59):10952-10968.
- Wang X, Li L, Shen X, Lu X. Rational Design Strategies in DNA-Encoded Libraries for Drug Discovery. Angew Chem Int Ed. 2025;64(34):e202511839.
- Abel R, Wang L, Harder ED, Berne BJ, Friesner RA. Advancing Drug Discovery through Enhanced Free Energy Calculations. Acc Chem Res. 2017;50(7):1625-1632.
- de Oliveira C, Leswing K, Feng S, Kanters R, Abel R, Bhat S. FEP Protocol Builder: Optimization of Free Energy Perturbation Protocols Using Active Learning. J Chem Inf Model. 2023;63(17):5592-5603.
- Moraca F, Negri A, de Oliveira C, Abel R. Application of Free Energy Perturbation (FEP+) to Understanding Ligand Selectivity: A Case Study to Assess Selectivity Between Pairs of Phosphodiesterases (PDE’s). J Chem Inf Model. 2019;59(6):2729-2740.
- Loeffler HH, He J, Tibo A, et al. Reinvent 4: Modern AI driven generative molecule design. J Cheminformatics. 2024;16(1):20.
- Moon SW, Ahn SH, Ahn JH, Kim HW. R2GB-GA: A Reaction-Regulated Graph-Based Genetic Algorithm for Exploring Synthesizable Chemical Space. Published online 2025.
- Parrot M, Tajmouati H, da Silva VBR, et al. Integrating synthetic accessibility with AI-based generative drug design. J Cheminformatics. 2023;15(1):83.
- Zhang X, Liu J, Xu B, et al. SynFrag: Synthetic Accessibility Predictor based on Fragment Assembly Generation in Drug Discovery. Published online 2025.
- Katsuyama A, Tokodai Y, Hoshi K, Ichikawa S. Retrosynthetic Analysis-Aware Fragment Linking for Structure-Guided Ligand Extension. Published online 2025.
- Khater T, Alkhatib SA, AlShehhi A, et al. Generative artificial intelligence based models optimization towards molecule design enhancement. J Cheminformatics. 2025;17(1):116.
- van den Broek RL, Patel S, van Westen GJP, Jespers W, Sherman W. In Search of Beautiful Molecules: A Perspective on Generative Modeling for Drug Design. J Chem Inf Model. 2025;65(18):9383-9397.
- Meyers J, Fabian B, Brown N. De novo molecular design and generative models. Drug Discov Today. 2021;26(11):2707-2715.
- Basak S, Bandyopadhyay A. Toward Making Polymer Chemistry Autonomous. ACS Appl Eng Mater. 2024;2(5):1190-1208.
- Struble TJ, Alvarez JC, Brown SP, et al. Current and Future Roles of Artificial Intelligence in Medicinal Chemistry Synthesis. J Med Chem. 2020;63(16):8667-8682.
- Wang G, Ang HT, Dubbaka SR, O’Neill P, Wu J. Multistep automated synthesis of pharmaceuticals. Trends Chem. 2023;5(6):432-445.
- Ali RSAE, Meng J, Jiang X. Synergy of Machine Learning and High-Throughput Experimentation: A Road Toward Autonomous Synthesis. Chem Asian J. 2025;20(20):e00825.
- Zhong L, Xu Y, Li X, Cheng P, Tao S. Production and evaluation of high-throughput reaction data from an automated chemical synthesis platform. Sci China Chem. 2025;68(10):5322-5331.
- Eyke NS, Koscher BA, Jensen KF. Toward Machine Learning-Enhanced High-Throughput Experimentation. Trends Chem. 2021;3(2):120-132.
- Mennen SM, Alhambra C, Allen CL, et al. The Evolution of High-Throughput Experimentation in Pharmaceutical Development and Perspectives on the Future. Org Process Res Dev. 2019;23(6):1213-1242.
- Xiao W, Su H, Ma Y, et al. Integration of Machine Learning and Automated Synthesis for Accelerated Drug and Material Research. ChemistrySelect. 2025;10(42):e04970.
- Liang X, Ren H, Han F, Liang R, Zhao J, Liu H. The new direction of drug development: Degradation of undruggable targets through targeting chimera technology. Med Res Rev. 2024;44(2):632-685.
- Baillie TA. Approaches to mitigate the risk of serious adverse reactions in covalent drug design. Expert Opin Drug Discov. 2021;16(3):275-287.
- Fan G, Chen S, Zhang Q, et al. Proteolysis-Targeting Chimera (PROTAC): Current Applications and Future Directions. MedComm. 2025;6(10):e70401.
- Vetma V, O’Connor S, Ciulli A. Development of PROTAC Degrader Drugs for Cancer. Annu Rev Cancer Biol. 2025;9(Volume 9, 2025):119-140.
- Sasso JM, Tenchov R, Wang D, Johnson LS, Wang X, Zhou QA. Molecular Glues: The Adhesive Connecting Targeted Protein Degradation to the Clinic. Biochemistry. 2023;62(3):601-623.
- Jung H, Lee Y. Targeting the Undruggable: Recent Progress in PROTAC-Induced Transcription Factor Degradation. Cancers. 2025;17(11):1871.
- Salama AKAA, Trkulja MV, Casanova E, Uras IZ. Targeted Protein Degradation: Clinical Advances in the Field of Oncology. Int J Mol Sci. 2022;23(23):15440.
- Baillie TA. Targeted Covalent Inhibitors for Drug Design. Angew Chem Int Ed. 2016;55(43):13408-13421.
- Péczka N, Orgován Z, Ábrányi-Balogh P, Keserű GM. Electrophilic warheads in covalent drug discovery: an overview. Expert Opin Drug Discov. 2022;17(4):413-422.
- Ray S, Murkin AS. New Electrophiles and Strategies for Mechanism-Based and Targeted Covalent Inhibitor Design. Biochemistry. 2019;58(52):5234-5244.