

Air Pollution’s Role in Alzheimer’s Disease Risk
Air Pollution as an Environmental Risk Factor for Alzheimer’s Disease and Related Dementias
Heui Hye Park1, Matthew J. Armstrong2, Fredric A. Gorin3 and Pamela J. Lein4
- Department of Molecular Biosciences, School of Veterinary Medicine, University of California, Davis, CA 95616, USA
- Department of Public Health, University of California, Davis, CA 95616, USA
- Department of Molecular Medicine, University of California, Davis, CA 95616, USA
- Department of Neurobiology, School of Medicine, University of California, Davis, CA 95616, USA
OPEN ACCESS
PUBLISHED: 31 October 2024
CITATION: PARK, Heui Hye et al. Air Pollution as an Environmental Risk Factor for Alzheimer’s Disease and Related Dementias. Medical Research Archives, [S.l.], v. 12, n. 10, nov. 2024. Available at: <https://esmed.org/MRA/mra/article/view/5825>. Date accessed: 22 nov. 2025. doi: https://doi.org/10.18103/mra.v12i10.5825.
COPYRIGHT: © 2025 European Society of Medicine. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
DOI: https://doi.org/10.18103/mra.v12i10.5825
ISSN 2375-1924
ABSTRACT
Alzheimer’s disease and related dementias are a leading cause of morbidity in our aging populations. Although influenced by genetic factors, fewer than 5% of Alzheimer’s disease and related dementia cases are due solely to genetic causes. There is growing scientific consensus that these dementias arise from complex gene by environment interactions. The 2020 Lancet Commission on dementia prevention, intervention, and care identified 12 modifiable risk factors of dementia, including lifestyle, educational background, comorbidities, and environmental exposures to environmental contaminants. In this review, we summarize the current understanding and data gaps regarding the role(s) of environmental pollutants in the etiology of Alzheimer’s disease and related dementias with a focus on air pollution. In addition to summarizing findings from epidemiological and experimental animal studies that link airborne exposures to environmental contaminants to increased risk and/or severity of Alzheimer’s disease and related dementias, we discuss currently hypothesized mechanism(s) underlying these associations, including peripheral inflammation, neuroinflammation and epigenetic changes. Key data gaps in this rapidly expanding investigative field and approaches for addressing these gaps are also addressed.
Keywords: Alzheimer’s disease, air pollution, environmental risk factors, dementia, neurodegeneration
Introduction
Alzheimer’s disease (AD) and AD-related dementia (ADRD) are global public health concerns. The worldwide prevalence of ADRD is projected to rise from 57.4 million affected individuals in 2019 to 152.8 million by 2050¹. Alzheimer’s disease and related dementias are the most prevalent type of progressive neurodegenerative disease that is pathologically characterized by amyloid beta plaques (Aβ), phosphorylated neurofibrillary tau tangles (NFT), and persistent neuroinflammation²⁵.
Clinical studies have identified that AD pathology develops years and often decades prior to the onset of AD symptoms⁶⁷. Mild cognitive impairment is a transitional phase of cognitive decline that progresses to dementia in approximately 30% of cases⁸⁹. Currently, there are no effective preventative or therapeutic interventions that effectively mitigate AD and ADRD risks or that prevent the advancement of AD. Given the increasing awareness of the burden of AD/ADRD, particularly as more family studies now support gene × environment (G × E) interactions, significant research effort is being devoted to identifying environmental risk factors for AD/ADRD and understanding how they modify disease risk and/or severity¹⁰⁻¹². This is because currently it is much easier to modify our environment than our genes.
Heterogeneity of Alzheimer’s Disease and Related Dementias
Alzheimer’s disease was initially described as a presenile dementia occurring in individuals between 45 and 65 years old, but in 1977 it was determined that the neuropathological findings of amyloid beta (Aβ) plaques and neurofibrillary tangles (NFTs), which are the hallmark pathologies of AD, were very similar in AD of early- and late-onset. However, it became increasingly recognized that with aging, late-onset AD brains frequently exhibited additional neuropathological findings, and these became categorized as ADRD. Before discussing environmental and genetic risk factors that contribute to AD and ADRD, it is important to explain terminologies used in this field.
Dementia describes a range of neurological conditions causing progressive deterioration of cognition and can be accompanied by additional neurological dysfunction. Alzheimer’s disease and related dementias are distinguished from other dementing illness by clinical assessment, neuroradiology, neuropathology, and biomarkers. Clinical assessment based on neurological and cognitive functional testing is least sensitive and least accurate at early stages of AD, and clinical confirmation utilizes other diagnostic tools including brain imaging modalities and blood and/or cerebrospinal fluid biomarker testing for amyloid and phosphorylated tau isoforms¹³.
The current evolution of increasingly selective and sensitive biomarkers for AD is necessary to accurately distinguish AD from other dementias and to evaluate treatments for AD/ADRD whose outcomes are dependent on disease stage or symptomatology¹⁴. Biomarker selectivity that distinguishes between the different types of ADRD will be essential to evaluate the interplay of genetic and other specific risk factors, but currently consensus has not been reached on future AD/ADRD classifications. For this review, we are summarizing prior literature based on current AD/ADRD classifications and guidelines.
In the current U.S. National plan to address AD, ADRD includes frontotemporal dementia, Lewy body dementia, vascular contributions to cognitive impairment and dementia, and mixed dementias (including cerebrovascular disease or Lewy bodies)¹⁵. Each of these dementias share progressive neuronal loss, cognitive and behavioral decline, impaired daily function, and aberrant protein accumulation (e.g., Aβ, tau, or α-synuclein)¹⁶¹⁷.
The heterogeneity of AD/ADRD complicates the task of identifying the interplay between specific genetic and environmental risk factors. While it has been easier to include ADRDs with AD when identifying environmental risk factors, it is acknowledged that this inclusive categorization increases the likelihood of “masking” specific G × E interactions that drive individual risk for specific forms of AD/ADRD.
GENETICS OF EARLY AND LATE ONSET ALZHEIMER’S DISEASE
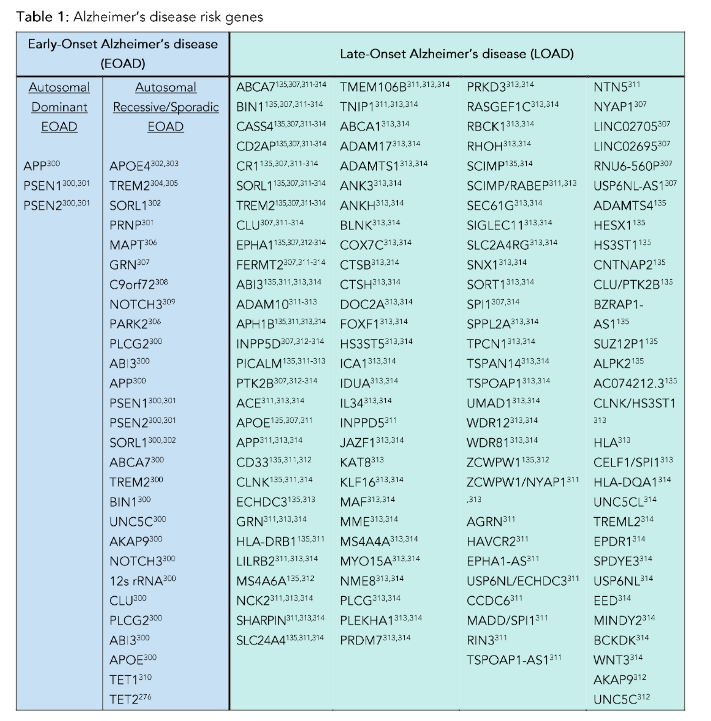
Broadly speaking, there are two age-based classifications of AD: early-onset Alzheimer’s disease (EOAD) and late-onset Alzheimer’s disease (LOAD). Early-onset Alzheimer’s disease is a less common form of AD diagnosed in individuals under the age of 65, often in their 40s and 50s. Late-onset Alzheimer’s disease, which is the more common form of AD, is diagnosed after age 65 and frequently includes additional neuropathology. While both forms of AD are highly heritable (~92–100%, and ~70–80%, respectively), they differ in their inheritance pattern¹⁸,¹⁹. Roughly 90% of EOAD is inherited in an autosomal recessive fashion, and 10% follows an autosomal dominant inheritance pattern. Additional subclassifications for EOAD have emerged, including Mendelian, non-Mendelian, or sporadic EOAD; however, in many studies of EOAD, the inheritance patterns are not delineated¹⁹.
Common autosomal dominant forms of EOAD are due to mutations in APP, PSEN1, and PSEN2, and autosomal recessive AD is linked to mutations in approximately 27 genes, including APOE4, BIN1, TREM2, MAPT, APP, UNC5C, and CLU (Table 1). While genome-wide association studies (GWAS) have identified multiple genes associated with increased risk of AD, the relationship of many of these risk loci to pathogenic mechanisms that drive disease progression have yet to be elucidated. Importantly, the differing clinical profiles of individuals carrying these high-risk alleles is consistent with a role for environmental factors interacting with genetic risks to determine individual outcomes.
The genetic contributions to LOAD are significantly more complex and include roughly 117 associated genes (Table 1). Overall, the heritability of LOAD is less than that of EOAD and findings from twin studies indicate a greater contribution of environmental factors in the development and progression of disease. A significant number of individuals with LOAD have co-morbid medical conditions such as cardiovascular disease, type 2 diabetes, autoimmune disorders²⁰–²¹, or head trauma²², each of which is separately associated with cognitive decline and each of which is heavily influenced by environmental factors. In addition to age-dependent increases in Aβ plaques and NFT, as many as 50% of LOAD brains contain pathologies of cerebral amyloid angiopathy, TDP-43 inclusions (LATE-NC) and/or Lewy body pathology (α-synuclein), which increase with age and AD progression²³. The presence of cerebrovascular cerebral amyloid angiopathy correlates with both Aβ plaques and NFT and is associated with expression of the APOE e4 allele independent of dementia status. Cerebral amyloid angiopathy, like AD, is a consequence of Aβ accumulation, but the intracerebral vascular accumulation produces a separate disorder with increased risk of stroke, cerebral hemorrhages, and inflammatory encephalopathies²⁴,²⁵ with its own distinct genetic risk factors²⁶ and diagnostic and potential clinical therapeutics. Given the modest risks conferred by each of these genes, as well as the co-morbid and environmental contributions to ADRD, it has been argued that LOAD, as well as most of ADRD, are driven by multifactorial influences, including multiple genes interacting with diverse environmental factors. Interestingly, many of the pathogenic mechanisms associated with these genetic risk alleles have been shown to be independently modified by environmental factors.
GENE BY ENVIRONMENT INTERACTION IN ALZHEIMER’S DISEASE AND RELATED DEMENTIAS ETIOLOGY
Twin studies have provided further evidence in support of the contribution of environmental factors to AD/ADRD etiology²⁷,²⁸. Specifically, genetically identical monozygotic twins have been compared to explore the effects of non-shared environmental risk factors. In one twin study, positron emission tomography (PET) imaging to quantify tau deposition in the entorhinal cortex and neocortical brain regions revealed similar intensity and deposition of tau pathology²⁷, suggesting that genetics was the predominant driver. However, when non-shared environmental risk factors were compared in twin pairs, differences in tau deposition strongly correlated with differences in exercise behaviors, social isolation, and physical inactivity²⁷. Similarly, another study of monozygotic twins showed that individuals exposed to higher levels of air pollution had lower structural integrity of the locus coeruleus, which is a brain region involved in the early stages of AD²⁸. These results suggest that while similarities in the development of AD-relevant pathologies between monozygotic twin pairs may be explained by identical genetic composition, different trajectories of ADRD pathologies may arise from exposure to non-shared environmental factors. In a different study, analysis of the AD polygenic risk score in monozygotic and dizygotic twins suggested that total genetic contribution to AD risk accounted for 71%, while the remaining 29% was attributed to environmental factors²⁹. This finding can help explain cases of discordant development of ADRD pathologies in monozygotic twins despite their genetic uniformity³⁰ and highlights the importance of identifying and studying the impact of environmental risk factors that can modify ADRD outcomes (Figure 1).
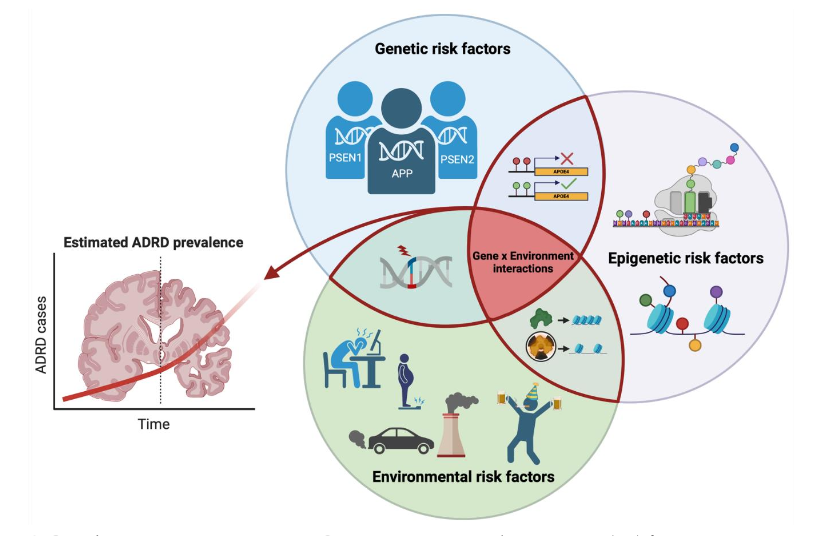
Figure 1. Gene by environment interactions.
Genetic, epigenetic, and environmental risk factors interact to promote ADRD etiology and disease progression.
NOTE: ADRD, Alzheimer’s disease-related dementias.
Created with BioRender.com.
Environmental risk factors for Alzheimer’s disease and related dementias
The 2020 Lancet Commission on dementia prevention, intervention, and care identified 12 modifiable environmental AD/ADRD risk factors based on meta-analyses and systemic reviews of the available literature¹². The report highlighted that these 12 modifiable risk factors contributed to 40% of global prevalence of dementia, suggesting that 40% of dementia cases worldwide might potentially be prevented or slowed by modifying individual exposures¹². The risk factors identified by the Lancet Commission included various comorbid medical conditions (hypertension, diabetes, hearing impairment, obesity, depression, and traumatic brain injury), lifestyles (physical inactivity, excessive alcohol consumption, social isolation, smoking), less educational background, and exposure to air pollution. Medical and public health strategies targeting comorbidity or lifestyles heavily depend on individual motivation, and these applications have been limited in success at the population level³¹. Many of these factors are strongly influenced by environmental contaminants, and environmental exposures represent a class of risk factors that can be changed by public policy, and thus have population-level impacts³¹.The contribution of environmental contaminants to the development of neurodegenerative diseases has been primarily studied in the context of occupational exposure to pesticides among agricultural workers.³² Individuals occupationally exposed to pesticides, such as organophosphate insecticides³³–³⁵ and fungicides that contain manganese³⁶–³⁸, were reported to have lower cognitive function and increased risk of developing neurodegenerative diseases, including AD³³,³⁹. Other synthetic persistent organic pollutants like per- and polyfluoroalkyl substances (PFAS) have also been associated with increased AD-relevant pathology and cognitive impairment in ADRD patients⁴⁰. More recently, air pollution (Box 1) has garnered significant interest due to increasing evidence from epidemiological and experimental animal studies of a strong association between air pollution and AD/ADRD. Additionally, while air pollution is directly linked to increased risk for AD/ADRD, it is also linked to a number of other ADRD risk factors, such as cardiovascular disease⁴¹–⁴⁶, metabolic dysfunction⁴⁷, and physical inactivity⁴⁸. The links between air pollution, AD/ADRD, and other risk factors suggest that air pollution may be an upstream environmental modifier that influences AD/ADRD onset and progression. It has been argued that targeting causative upstream environmental modifiers will have a broader and more significant impact on reducing the AD burden than modifying proximal individual-level risk factors⁴⁹. This review focuses on current understanding and findings regarding the association between air pollution and increased risk and/or severity of AD/ADRD and discusses prevailing hypotheses regarding the mechanisms underlying these links.Box 1. Air pollution composition, toxicology and regulations

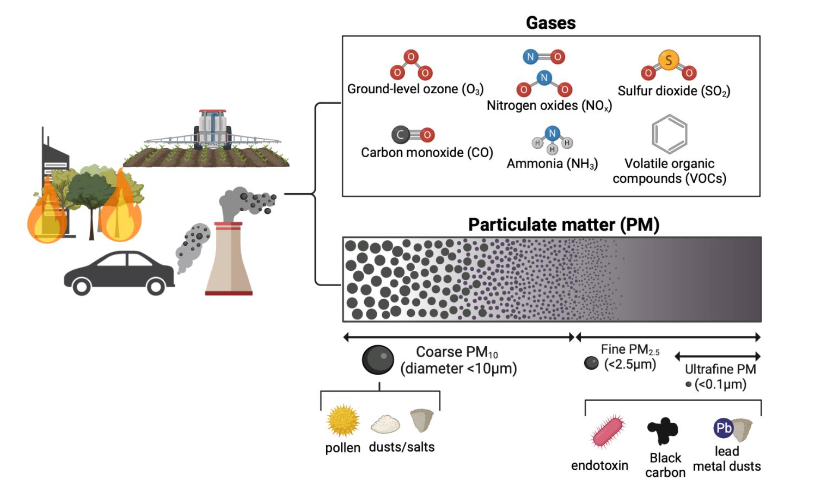
Figure 2. Composition of air pollutants.
Air pollutants consist of gaseous and particulate matter (PM) fractions with heterogeneous sources, compositions, and sizes, all of which influence toxicity to the respiratory system and the brain.
Created with BioRender.com.
Epidemiological evidence linking air pollution with progressive cognitive impairment and Alzheimer’s disease and related dementias
Over 80 epidemiological studies investigating an association between air pollution and AD/ADRD were published in the peer-reviewed literature during the last six years (Table 2). Recent studies have focused on correlations between independent and combined effects of specific air pollutants and AD/ADRD risk. Zip code-based residential exposure levels of fine and course particulate matter (PM₂.₅, PM₁₀), nitrogen oxides (NOₓ), and ground-level ozone (O₃), have been leveraged to calculate odds ratios or hazard ratios to assess the risk for AD/ADRD conferred by exposure to air pollutants, with increasing risk associated with increasing levels of each air pollutant.
Longitudinal monitoring studies of average air pollution exposure levels have also been leveraged to measure the time-length effects of long-term exposure to air pollution on cognitive decline and AD/ADRD⁵⁰–⁵⁴.
Population-based studies (n = 150–8,000) to nationwide studies (n = 4–12 million) of midlife to aged participants (age > 45). The main ADRD outcomes that were measured against air pollution exposure levels were risks of incident ADRD determined by clinical records of diagnosis, hospitalization, pathology of AD-relevant biomarkers, cognitive assessment, and mortality.
Table 2: Summary of epidemiological studies testing the association between air pollutants and Alzheimer’s disease-related dementias
| Air pollutant | AD-relevant clinical pathology | Cognitive impairment | ADRD risk |
|---|---|---|---|
| PM₂.₅ | ↑ 5 studies reported positive association⁵³,⁵⁴,¹⁰⁴,¹⁰⁶,¹¹¹ ⦸ 1 study reported no association¹⁰⁵ |
↑ 13 studies reported positive association⁵¹,⁸¹,¹⁰⁵,¹¹⁵–¹²¹,¹²⁶,¹³⁴,³¹⁵ ⦸ 4 studies reported no association⁹⁹,¹⁰³,¹¹⁴,¹²⁵ |
↑ 36 studies reported positive association⁵⁰–⁵²,⁵⁸–⁹⁰ ⦸ 5 studies reported no association⁹⁵–⁹⁹ |
| PM₁₀ | ↑ 4 studies reported positive association⁵⁴,¹⁰⁴,¹⁰⁶ | ↑ 4 studies reported positive association¹⁰⁶,¹²¹,¹²²,¹³⁴ ⦸ 3 studies reported no association⁹⁹,¹⁰³,¹²⁵ |
↑ 9 studies reported positive association⁴⁶,⁷⁰–⁷²,⁸⁰,⁸²,⁸⁵,⁸⁷,¹⁰⁷ ⦸ 1 study reported no association⁷⁸ |
| NO₂ | ↑ 4 studies reported positive association⁵⁴,¹⁰⁴,¹⁰⁶ | ↑ 6 studies reported positive association¹⁰⁶,¹¹⁸,¹²¹,¹²³,¹³⁴,³¹⁵ ⦸ 4 studies reported no association⁹⁸,¹⁰³,¹¹⁴,¹²⁵ |
↑ 28 studies reported positive association⁵⁰,⁵⁸,⁵⁹,⁶¹–⁶³,⁶⁶,⁷⁶,⁷⁹–⁸³,⁸⁶–⁸⁷,⁹³–⁹⁵ ⦸ 8 studies reported no association⁵²,⁷⁵,⁷⁹,⁸⁴–⁹⁵ |
| O₃ | ↑ 1 study reported positive association¹²⁰ ⦸ 1 study reported no association⁵³ |
↑ 1 study reported positive association¹²⁰ ⦸ 1 study reported no association¹²³ |
↑ 3 studies reported positive association⁵⁸,⁸²,⁸³ ⦸ 6 studies reported no association⁵³,⁶⁸,⁷⁶,⁸⁷,⁹⁴,⁹⁵ |
| Black carbon | — | ↑ 4 studies reported positive association⁶⁷,⁷⁶,⁹⁷,⁹⁸ ⦸ 4 studies reported no association⁶²,⁷⁰,⁹⁴,⁹⁷,⁹⁹ |
— |
NOTE:
AD, Alzheimer’s Disease; ADRD, Alzheimer’s disease-related dementias; AD-relevant clinical pathology includes Aβ42/40 and neurofilament light levels in the blood and cerebrospinal fluid, and brain PET/MRI scans for amyloid, tau, and cortical thickness detection.
↑: positive association reported; ⦸: no association reported.
SUMMARY OF FINDINGS
In line with earlier epidemiology studies⁵⁵–⁵⁷, the main consensus of current epidemiological findings is that there is a strong association between exposure to air pollution and AD/ADRD; however, there were mixed results regarding associations between air pollutant type and increased risk of AD/ADRD. The majority of studies found a positive correlation between risk of all-cause dementia and exposure to PM₂.₅⁵⁰–⁵²,⁵⁸–⁹⁰ and NO₂⁵⁰,⁵⁸,⁵⁹,⁶¹–⁶³,⁶⁶,⁶⁷,⁹³–⁹⁵,⁸⁰–⁸²,⁸⁷–⁹⁷,³⁰³ but not to O₃⁵³,⁶⁸,⁷⁶,⁸⁷,⁹⁴,⁹⁵ (Table 2). A few studies reported no significant association between risk of dementia and levels of PM₂.₅⁹⁵–⁹⁹ and NO₂⁵²,⁷⁵,⁹⁴–⁹⁵, and a small number of studies found a positive association between O₃ level and risk of dementia⁵⁸,⁸²,⁹³.
When records of dementia diagnoses are more specifically stratified, differential correlations between various types of air pollutants and subtypes of dementia emerge. In an analysis of data from the UK Biobank, authors reported that exposure to combined levels of NO₂, PM₂.₅, and PM₁₀ were significantly associated with all-cause dementia, AD, vascular dementia (VaD), and mild cognitive impairment. Yet, when each air pollutant was compared separately, VaD was no longer associated with PM₂.₅¹⁵⁰ but only influenced by NO₂, while all-cause dementia, AD, and mild cognitive impairment remained significantly associated with both PM₂.₅, PM₁₀, and NO₂⁸³.
Another study leveraging the UK Biobank data found similar associations of PM₂.₅, PM₁₀, and NO₂ with all-cause dementia and AD.
but not with VaD⁶⁶. In line with these findings, other studies reported the association of PM₂.₅ to be more pronounced with AD than with non-AD or VaD⁶¹,⁶⁴,⁶⁵,⁷⁴, but contrary results showed more significant association with non-AD or VaD than with AD²⁸,⁸⁴,¹⁰⁰. These mixed findings may reflect differences across studies in modeling methods, cohort characteristics, exposure duration, confounding co-exposures, source of air pollution data, and dementia ascertainments. Due to the heterogeneity of dementia etiology and disease progression, inconsistent clinical diagnoses and records may have been used across different studies with participants from a wide range of cohorts. Additionally, the outcome measurements mainly derived from medical records may not be an accurate representation of the entire population because of underlying socioeconomic disparities in access and accessibility of medical care¹⁰². This could be a critical confounding factor because studies have shown that social disadvantage can greatly enhance pollution-mediated risk of AD/ADRD¹⁰¹,¹⁰³.
Despite these mixed results, long-term exposure to higher levels of PM₂.₅ and NO₂ was still consistently found to be associated with increased risk of AD and ADRD in most studies (Table 3). Long-term exposure to higher levels of PM₂.₅ and NO₂ was associated with greater extent of AD-relevant structural changes in the brain, which were assessed using PET and magnetic resonance imaging (MRI) to quantify amyloid deposition and cortical atrophy²⁸,⁵³,¹⁰⁴–¹⁰⁷. Individuals who were exposed to higher levels of PM₂.₅ and NO₂ exhibited higher amyloid PET positivity and cortical atrophy⁵³,¹⁰⁴–¹⁰⁷. In smaller cohorts, AD-relevant biomarkers, such as Aβ 42/40 and neurofilament light levels in the plasma and cerebrospinal fluid, were positively associated with PM₂.₅ and NO₂ exposure levels⁵⁴,¹⁰⁴,¹⁰⁸. There were a few post-mortem autopsy studies that reported increased expression of histological biomarkers of AD/ADRD, including hyperphosphorylated tau, Aβ deposition, nanoparticle inclusions, and glial activation in the hippocampus, cortex, and olfactory bulbs of young and old individuals who lived in areas with high levels of air pollution¹⁰⁹–¹¹³.
Another common outcome measurement was cognitive performance, which was assessed by a battery of cognitive tests to assess different cognitive domains and executive functions¹¹⁴–¹¹⁸. While some studies reported an inverse correlation between cognitive test scores and exposure to PM₂.₅ and NO₂ in cognitively unimpaired and impaired populations¹⁰⁶,¹¹⁵–¹²⁴, other studies found the relationship to be not significant⁹⁸,¹⁰⁵,¹¹⁴,¹²⁵.
Table 3: Epidemiological studies studying air pollution as a risk factor for Alzheimer’s disease-related dementias
| Air pollutant levels | Participant follow-up duration | Study results | Cohort type/size | Citation / location |
|---|---|---|---|---|
| Annual mean: PM₂.₅ = 9.3 μg/m³ NO₂ = 17.1 ppb O₃ = 42.6 ppb |
2000–2018 | PM₂.₅, NO₂ ↑ incident AD/ADRD O₃ ⦸ incident AD/ADRD |
Medicare Chronic Conditions Warehouse Age ≥ 65 yrs n = 12,233,371 |
Shi, et al.⁵⁸ United States |
| Annual median: PM₂.₅ = 10.6 μg/m³ NO₂ = 18.26 ppb O₃ = 46.68 ppb |
2000–2016 | PM₂.₅, NO₂ exposure 8–10 yrs prior to diagnosis ↑ first ADRD hospitalization | Medicare FFS beneficiaries Age ≥ 65 yrs n = 8,507,437 |
Mork, et al.⁵⁰ United States |
| 2010 median: PM₂.₅ = 10 μg/m³ PM₁₀ = 16.1 μg/m³ NO₂ = 43.4 μg/m³ |
Median 12.01 yrs | PM₂.₅, NO₂ ↑ all-cause dementia risk NO₂ ↑ AD risk in individuals with higher genetic risk scores |
UK Biobank Study Age ≥ 50 yrs n = 437,932 |
Yuan, et al.⁵⁹ United Kingdom |
| Air pollutant levels | Participant follow-up duration | Study results | Cohort type/size | Citation/location |
|---|---|---|---|---|
| 10-year median: PM₂.₅ = 11.2 μg/m³ |
1998–2016 | PM₂.₅ ↑ incident dementia | Health and Retirement Study Age > 50 yrs n = 27,857 |
Zhang, et al.⁵¹ United States |
| 10-year median: PM₂.₅ = 21.3 μg/m³ NO₂ = 32.8 μg/m³ BC = 2.3×10⁻⁵/m |
1999–2011 | PM₂.₅ ↑ all-cause dementia, AD, and VaD risks NO₂, BC ⦸ dementia risk |
Three-City Study Age ≥ 65 yrs n = 7,066 |
Mortamais, et al.⁵² France |
| Annual mean: TRAP PM₂.₅ = 0.18 μg/m³ Wood-burning PM₂.₅ = 0.77 μg/m³ |
1993–2010 | All sources of PM₂.₅ ⦸ dementia incidence | Betula Study Age > 55 yrs n = 1,806 |
Oudin, et al.⁶⁰ Sweden |
| Biennial median PM₂.₅ range: 2002–2003: 10.09–12.65 μg/m³ 2015–2016: 7.4–8.34 μg/m³ |
2016–2018 | PM₂.₅ ↑ amyloid PET scan positivity O₃ ⦸ amyloid PET scan positivity |
IDEAS Study (Imaging Dementia–Evidence For Amyloid Scanning) Age ≥ 65 yrs n = 18,178 |
Iaccarino, et al.⁵³ United States |
| 1- to 20-year average: PM₂.₅ = 5–25 μg/m³ PM₁₀ = 10–40 μg/m³ NO₂ = 5–40 ppb |
2000–2008 | PM₂.₅, PM₁₀, NO₂ ↑ plasma Aβ1-40, Aβ1-42, Aβ1-42/1-40 ratios | Ginkgo Evaluation of Memory Study Age ≥ 75 yrs n = 3,034 |
Hajat, et al.⁵⁴ United States |
| 5-year mean: PM₂.₅ = 25.9 μg/m³ PM₁₀ = 49.7 μg/m³ NO₂ = 27.0 ppb |
2014–2017 | PM₁₀, NO₂ ⦸ MRI AD-like cortical atrophy PM₂.₅ ⦸ MRI AD-like cortical atrophy PM₂.₅, PM₁₀, NO₂ ↑ MRI non-AD-like cortical atrophy PM₁₀, NO₂ ⦸ MoCA cognitive score |
EPINEF Study (Environmental Pollution-Induced Neurological Effects) Age ≥ 50 yrs n = 640 |
Cho, et al.¹⁰⁶ Korea |
| Annual mean: PM₂.₅ = 17.2 μg/m³ PM₁₀ = 37.7 μg/m³ NO₂ = 57.3 μg/m³ NOₓ = 99.1 μg/m³ |
2013–2014 | PM₁₀, NO₂ ⦸ MRI cortical thickness PM₂.₅ ⦸ MRI cortical thickness PM₂.₅, PM₁₀, NO₂ ⦸ cognitive function |
ALFA+ Study (Alzheimer and Families) Cognitively unimpaired adults Age 45–75 yrs n = 958 (cognition) n = 228 (MRI) |
Crous-Bou, et al.¹⁰⁵ Spain |
| Annual mean: PM₂.₅ = 17.3 μg/m³ PM₁₀ = 37.9 μg/m³ NO₂ = 57.6 μg/m³ |
2013–2014 | PM₂.₅, NO₂ ↑ PET amyloid PM₂.₅, PM₁₀ ↑ CSF NFL stronger in APOE-ε4 carriers |
ALFA+ Study Cognitively unimpaired adults Age 57 yrs n = 156 |
Alemany, et al.¹⁰⁴ Spain |
LIMITATIONS
All epidemiological studies are limited by several factors. In the studies described, synergistic effects between air pollution and other genetic⁵¹,²⁶, lifestyle⁸⁷,¹⁰¹,¹²²,¹²⁶–¹²⁹, and environmental⁵⁷,⁵⁸,¹⁰¹,¹⁰³,¹⁰⁵,¹²⁸,¹³⁰–¹³³ risk factors may not have been completely adjusted for in the analyses. Although carrying the APOE ε4 gene is a significant AD risk factor¹³⁴–¹³⁶, results from studies assessing whether it has a modifying effect on the association between air pollution and AD are inconclusive: some showed the AD/ADRD risk associated with air pollution exposure to be more pronounced in APOE ε4 carriers⁵⁷,⁷²,¹⁰⁴,¹¹⁶,¹¹⁹,¹²¹, while others did not⁴⁶,⁷²,⁷³,⁷⁷,¹⁰⁴,¹⁰⁵.
Genetic susceptibilities other than APOE ε4 status that can change the trajectory of AD pathogenesis were not taken into consideration for most studies. Moreover, other lifestyle factors, such as physical activity, exposure to traffic noise, sleeping patterns, and socioeconomic status, all of which are known to affect AD development⁸⁷,¹²⁷,¹³⁷–¹⁴⁰ may not have been fully adjusted for in the studies. Since the G × E interactions that lead to AD/ADRD progression are complicated and not well understood, it is challenging for epidemiological studies to fully capture the true impact of air pollution on AD/ADRD. Nevertheless, consistent epidemiological findings across different cohorts and countries that show a robust association between air pollution and ADRD provide strong justification for further experimental investigation of the underlying mechanisms by which air pollution increases AD/ADRD risk.
Experimental animal studies: The challenge of replicating human exposures to air pollution
To investigate the impact of air pollution on AD/ADRD pathogenesis, experimental animal studies have primarily used wildtype or transgenic rodent models that express human AD risk genes to determine whether exposure to air pollution decreases the time to onset or the severity of AD-relevant phenotypes.
A major challenge of studying air pollution toxicology in an experimental animal model is replicating human-relevant exposures to air pollution¹⁴¹,¹⁴². Most earlier studies employed acute exposures to high concentrations of diesel exhaust particles (DEP) or PM₂.₅ administered to rodents via intratracheal¹⁴³ or intranasal¹⁴⁴ instillation, or, alternatively, oropharyngeal aspiration¹⁴⁵ of ambient PM₂.₅ samples extracted from filtered media and resuspended in a delivery vehicle. The advantages of these instillation or aspiration exposure routes are that the composition of particles, dosage, and site of delivery are easier to control in testing the toxicity of a known amount and composition of air pollutants in a cost-effective way¹⁴⁶.
However, there are several critical shortcomings of these exposure paradigms in terms of replicating human exposure, including:
(1) inaccurate reproduction of inhaled particle deposition,
(2) lack of exposure to gaseous co-pollutants¹⁴⁶.
These factors can influence the toxicity of air pollutants and induce inconsistent biological responses even to the same air pollutants¹⁴²,¹⁴⁷,¹⁴⁸. Hence, many recent studies have utilized in vivo whole-body inhalation exposures to aerosols of air pollutants to test toxicity in a manner that more closely replicates human exposure¹⁴⁹–¹⁶¹. Some studies exposed animals to laboratory-generated aerosols of concentrated PM₂.₅, UFPM, or O₃¹⁴⁹–¹⁵⁴, while other studies implemented variations of concentrated ambient particle (CAP) systems to collect real-world ambient air and concentrate PM₂.₅ or UFPM for direct delivery to animals¹⁵⁵–¹⁵⁸. The CAP systems allow more accurate representation of real-world compositions of PM, yet because CAPs methodology only concentrates particles and not gases, understanding combined effects of PM and gases on biological outcomes is not possible¹⁴¹,¹⁵⁹. Lastly, both laboratory-generated and CAP-generated aerosols do not effectively emulate chronic real-life exposure dynamics, which vary hourly, weekly, monthly and seasonally¹⁴¹.
A few in vivo studies have developed methods for collecting ambient polluted air and delivering it to animals unchanged and in real-time at ambient concentrations via whole-body inhalation¹⁴¹,¹⁶⁰–¹⁶². For example, in one study, ambient traffic-related air pollution (TRAP) was collected from a heavily trafficked freeway tunnel in northern California, USA, for subsequent delivery in real-time to animals housed in an adjacent vivarium¹⁴¹. This study demonstrated that chronic life-time exposure to ambient TRAP exacerbated AD-relevant phenotypes in wildtype Fischer rats and transgenic TgF344-AD rats that expressed human AD risk genes¹⁶⁰. Another study in Taipei, Taiwan used the Taipei Air Pollutant Exposure System (TAPES) to sample and deliver outdoor ambient air mixture directly to 3xTg-AD mice housed in an exposure chamber. Transgenic mice exposed to ambient air exhibited increased AD neuropathologies relative to filtered air controls¹⁶¹. These novel exposure paradigms in rodent models increase the representativeness of the exposure conditions and provide corroborative evidence to support the epidemiological findings.
Experimental animal studies: Chronic exposure to air pollution exacerbates Alzheimer’s disease pathology
A number of experimental animal studies have quantified Aβ plaques, NFT, tau phosphorylation, neuronal cell loss, and cognitive behavior deficits in both wildtype and transgenic rodent models after exposure to air pollution (Table 4). In most of these studies, the exposure paradigms were short-term or subchronic exposures to PM₂.₅ and UFPM that ranged from 2–13 weeks¹⁴⁹–¹⁵²,¹⁵⁵–¹⁶¹. Only a few studies examined the neurological impacts of chronic exposures, ranging from 5–14 months¹⁰⁹,¹⁵³,¹⁵⁴,¹⁶⁰,¹⁶².
Results from these studies are mixed depending on the exposure paradigm and animal model. Studies with more chronic exposure durations (Table 4) more consistently reported air pollution–induced increases in Aβ/NFT loads, tau hyperphosphorylation, microgliosis, astrogliosis, and neuronal atrophy. Exposures longer than 3 months at either concentrated or ambient levels of UFPM, PM₂.₅, or O₃ were shown to increase Aβ plaque load, tau phosphorylation, gliosis, and worsen markers of neuroinflammation in the hippocampus, cortex, and olfactory bulbs of rodents¹⁴⁹,¹⁵⁰,¹⁵³,¹⁵⁷,¹⁵⁸,¹⁶⁰. Some short-term exposure studies (2–3 weeks) in rodents reported neuronal atrophy and memory deficits¹⁵³,¹⁵⁵,¹⁶³, while others did not observe those changes¹⁴⁹,¹⁶¹,¹⁶⁴ or observed contrary results of reduced Aβ plaques with increased microgliosis¹⁵⁶. The mixed results may be partially attributed to varying degrees of neuroinflammation caused by air pollution, as discussed below.
Table 4: Summaries of in vivo studies of the effects of air pollution on Alzheimer’s disease neuropathology
| Exposure system | Exposure paradigm | Animal model | Study results | Citation |
|---|---|---|---|---|
| System: Caldecott Tunnel Exposure Facility (CTEF) Source: TRAP drawn from tunnel bores in Oakland, CA, USA |
Whole-body exposure for 2, 5, 9, and 14 months (7 days/week, 24 h/day) PM₂.₅ = 15.6 ± 3.7 μg/m³ |
Male & female TgF344-AD & WT rats (Fischer344) 1-month-old |
↑ Refractive particle deposition in HIP after 5 months ↑ p-Tau in HIP after 14 months ↑ Aβ deposition in CO after 10 months ↑ Neuronal cell death in EC after 14 months ↑ Neuroinflammation in HIP, EC (microglia activation, IL-1β, TNF-α) ↓ Learning, memory after 14 months ⦸ Serum cytokine |
Patten, et al.¹⁶⁰,¹⁶² |
| System: Taipei Air Pollutant Exposure System (TAPES) |
Whole-body exposure for 3 months (7 days/week, 24 h/day) | Female 3xTg-AD mice 6-month-old |
↑ p-Tau, oxidative stress (MDA) in HIP, OB ↑ Neuronal cell death in EC ⦸ Aβ42 deposition, learning |
Lee, et al.¹⁶¹ |
| Exposure system | Exposure paradigm | Animal model | Study results | Citation |
|---|---|---|---|---|
| Source: Outdoor ambient air drawn from Taipei, Taiwan |
PM₂.₅ = 11.38 μg/m³ | — | — | — |
| System: Harvard ultrafine concentrated ambient particle system (HUCAPS) Source: TRAP UFPM drawn from Rochester, NY, USA |
Whole-body exposure for 2 weeks (4 days/week, 4 h/day) UFPM = 57 μg/m³ Mean UFPM size = 79 nm |
Male 3xTgAD & WT mice 12.5-month-old |
↓ Learning, memory ↓ Olfactory discrimination |
Jew, et al.¹⁵⁵ |
| System: Harvard ultrafine concentrated ambient particle system (HUCAPS) Source: Ambient UFPM drawn from street |
Whole-body exposure for 2 weeks (4 days/week, 4 h/day) UFPM = 42 ± 15.7 μg/m³ |
Male 3xTgAD & WT mice 12.5–14-month-old |
↑ p-Tau (pT205) in HIP ⦸ Aβ deposition in HIP ↓ Aβ deposition in subiculum ↑ Microglial ramification in HIP |
Herr, et al.¹⁵⁶ |
| System: Versatile Aerosol Concentration and Enrichment System (VACES) Source: TRAP UFPM drawn from Orange County, CA, USA |
Whole-body exposure for 12 weeks (4 days/week, 5 h/day) UFPM = 65.4 μg/m³ |
Male and female APPⁿˡᶜᵏ⁺/⁺;KI & WT mice (C57/BL6) 3-, 9-month-old |
↓ Learning, memory in both young and aged mice ↑ Aβ deposition in CO, HIP ↑ Neuroinflammation in CO only in old mice (microglia & astrocyte activation) ⦸ Neuronal atrophy |
Kilian, et al.¹⁵⁷ |
| System: Versatile Aerosol Concentration and Enrichment System (VACES) Source: PM₂.₅ from Columbus, OH, USA |
Whole-body exposure for 3 months (5 days/week, 6 h/day) PM₂.₅ = 25.8 μg/m³ |
Male APP/PS1 & WT mice (C57BL/6; C3H) 12-month-old |
↑ Neuroinflammation in HIP (microglia & astrocyte activation; TNF-α, IL-1β, IL-6, MIP-3α, IFN-γ) ↑ Aβ deposition, β-secretase, γ-secretase |
Sahu, et al.¹⁵⁸ |
| System: Chambers housed outdoor Source: Ambient outdoor air in Santiago, Chile PM₂.₅ ≥ 55 μg/m³ |
Whole-body exposure for 7 months (7 days/week, 24 h/day) | Female C57/BL6 mice 2-month-old |
↑ DNA double-strand break marker (γ-H2AX) ↑ p-Tau in CO |
Calderon-Garcidueñas, et al.¹⁰⁹ |
| System/source: Soot UFPM generated by a Combustion Aerosol Standard burner |
Whole-body exposure for 2 weeks (5 days/week, 4 h/day) UFPM = 7.0×10⁵ particles/cm³ Mean UFPM size = 39 nm |
Female 5xFAD & WT mice (C57BL/6J) 6-month-old |
↑ Inflammation (IFN-γ in plasma, OB, HIP) ↑ Oxidative stress in HIP (SOD2 protein) ⦸ Aβ deposition or microglial and astrocytic activation in HIP and CO |
Savelieva, et al.¹⁴⁹ |
| System/source: UFPM generated by a PM generator |
Whole-body exposure for 3 weeks (5 days/week, 8 h/day) UFPM = 1000 μg/m³ Mean UFPM size = 178 ± 65 nm |
Male WT mice (C58BL/6J) 12-month-old |
↑ Oxidative stress (4-HNE) in HIP ↑ Aβ deposition, neuroinflammation (TNF-α) in HIP ⦸ Neuronal cell death in HIP |
| Exposure system | Exposure paradigm | Animal model | Study results | Citation |
|---|---|---|---|---|
| System/source: O₃ generated by an HFL-10.0 O₃ generator |
Whole-body exposure for 13 weeks (3 days/week, 4 h/day) O₃ ≈ 1.0 ppm |
Male 5xFAD & WT mice (C57BL/6J) 10–11-week-old |
↑ Aβ deposition in HIP and CO ↓ Plaque-associated microglia ↓ TREM2 on plaque-associated microglia in CO ↑ Neuroinflammation in CO (NLRP3, IL-1β) ↑ Neurite dystrophy (TLAMPl, VAChT, ChAT, AChE) in CO |
Greve, et al.¹⁵⁰ |
| System/source: O₃ generated by an HFL-10.0 O₃ generator |
Whole-body exposure for 13 weeks (3 days/week, 4 h/day) O₃ ≈ 1.0 ppm |
Male 5xFAD mice (C57BL/6J) 10–11-week-old |
↑ Aβ deposition in CO ↑ Astrocyte, astrocyte–microglia interaction in peri-plaque regions ↑ Neuroinflammation in CO (DAM astrocytic profile) |
Ahmed, et al.¹⁵¹ |
| System/source: O₃ generated by a flux ozone generator |
Whole-body exposure for 7, 15, 30, 60, and 90 days (7 days/week, 4 h/day) O₃ = 0.25 ppm |
Male WT rats (Wistar) 250–300 g |
↑ Aβ structural destabilization over time (β-helix, β-sheet) ↑ Intracellular Aβ1-42 after 60–90 days ↑ Lipid peroxidation, microglial and astrocytic activation in HIP after 15–90 days ↓ Neuronal cell death in HIP after 90 days |
Rivas-Arancibia, et al.¹⁵³,¹⁵⁴ |
NOTE:
HIP = hippocampus, CO = cortex, EC = entorhinal cortex; p-Tau = phosphorylated tau; WT = wildtype; ↑ = increase in measurement; ↓ = decrease in measurement; ⦸ = no change in measurement.
Blue cells: pollutants from real-world sources. Yellow cells: pollutants generated in the laboratory.
Prevailing hypothesis: Air pollution promotes Alzheimer’s disease–related dementias phenotypes via inflammatory mechanisms
Chronic neuroinflammation in Alzheimer’s disease pathogenesis
Persistent neuroinflammation has been posited as a key mechanism responsible for progressive neurodegenerative diseases, including ADRD¹⁶⁵, based on accumulating evidence from human post-mortem¹⁶⁶–¹⁶⁸ and experimental animal studies¹⁶⁹,¹⁷⁰.
Aggregates of misfolded proteins like Aβ and NFT, which are neurotoxic, have been shown to trigger neuroinflammation¹⁷¹,¹⁷². Neuroinflammatory responses are mainly orchestrated by resident brain immune cells — microglia and astrocytes — that protect the brain microenvironment not only by phagocytosing cellular damage, but also by regulating neurogenesis and synapse number¹⁷³,¹⁷⁴. While microglia-mediated amyloid clearance¹⁶⁹,¹⁷⁵ and pruning of axosomatic inhibitory synapses after injuries¹⁷⁶,¹⁷⁷ likely provide neuroprotection, dysregulation of these microglial functions is observed in AD/ADRD, and an increasing number of studies report that pro-inflammatory or disease-associated microglial phenotypes are associated with worse neurotoxic outcomes and increased severity of AD/ADRD neuropathology¹⁶⁹,¹⁷⁸–¹⁸².
Exposure to air pollution promotes neuroinflammation
Glial activation in response to PM exposure has been quantified by immunohistochemical analyses in many experimental animal studies. Most studies used IBA-1 as a biomarker of microglia¹⁸³,¹⁸⁴ and GFAP as a biomarker of astrocytes¹⁸⁵,¹⁸⁶, and some studies colocalized IBA-1 with CD68, a marker of phagocytic cells.
Numerous studies have reported that PM induced recruitment of microglia and astrocytes to the olfactory bulb and hippocampus of rodents¹⁵⁸,¹⁶⁰,¹⁸⁷. However, PM-induced microglial activation varied across different studies depending on the exposure paradigm, sex, age, genotype, and brain region.
Mice exhibited increased IBA-1⁺ expression with more amoeboid microglial morphology in the olfactory bulb after 4–8 weeks of exposure to concentrated PM₂.₅¹⁸⁷, whereas in another study, APP/PS1 mice, but not wildtype mice, exhibited increased microglial activation in the hippocampus following a 3-month exposure¹⁵⁸. A more chronic exposure to ambient TRAP induced microglial phagocytosis (increased ratio of CD68⁺/IBA-1⁺) in the hippocampus of both wildtype and TgF344-AD female rats after 2 months of exposure, while male wildtype and TgF344-AD rats did not exhibit TRAP-induced microglial activation until after 14 months of exposure¹⁶⁰. Interestingly, in contrast to male rats, female rats exhibited a dampened microglial phagocytic response to TRAP after 14 months of exposure¹⁶⁰. A different study reported that 2 weeks of UFPM exposure reduced microglial recruitment with more ramified microglial morphology in 3xTgAD mice compared to their air-exposed controls¹⁵⁶.
Likewise, PM-induced astrocytic activation (GFAP⁺ immunoreactivity) varied across studies. While some studies reported PM-induced astrocytic recruitment and activation in the hippocampus¹⁵²,¹⁵⁸,¹⁸⁸, hypothalamus¹⁸⁹, and striatum¹⁹⁰, other studies reported no significant effects of PM on astrocytic activation in any of the brain regions examined¹⁵⁶,¹⁶⁰.
These variable observations suggest that the glial responses to air pollution are complex. The functional impact of these changes also is not clear since it is now appreciated that glial activation may either exacerbate¹⁶⁹,¹⁷²,¹⁷⁸–¹⁸² or protect¹⁶⁹,¹⁹¹–¹⁹³ against synapse loss and neurodegeneration. To better understand how air pollution disrupts neuroinflammatory homeostasis, more recent studies have distinguished glial phenotypes using molecular biomarkers of pro- vs. anti-inflammatory phenotypes or biomarkers of disease-associated microglia (DAM). Following 12 weeks of exposure to UFPM, TMEM119⁺ microglia, which are considered homeostatic, were not associated with amyloid plaques were reduced, while ferritin⁺ DAM microglia, which are associated with senescence and reactive oxygen species (ROS), were increased in both wildtype and App^NL-G-F^+/KI mice¹⁵⁷. The same study also showed that UFPM increased the number of highly inflammatory, neurotoxic C3⁺ DAM astrocytes in the cortex of both genotypes¹⁵⁷. In vitro studies also have characterized glial phenotypes after PM treatment. Primary coculture of microglia and astrocytes exhibited upregulated expression of DAM astrocytic markers (C3 protein and DAM-specific transcripts) in response to PM₂.₅ treatment¹⁹⁴. In addition, PM₂.₅ altered interactions between neurons and glial cells in a triculture of neuron, astrocyte, and microglia human cell lines by promoting polarization of DAM microglial characterized by CD11b, CD68, and iNOS expression¹⁹⁵. Based on these results, the type of glial activation or polarization following PM exposure may be important in either resolving inflammatory signaling or tilting it towards persistent neuroinflammation and glial dysfunction, which can further exacerbate AD pathogenesis.
These observations raise important questions regarding the mechanism(s) by which air pollutants alter glial phenotypes. Microglia and astrocytes are activated by autocrine and paracrine signals in the forms of:
(1) cytokines and chemokines, which are small immune signaling molecules¹⁹⁶, and
(2) reactive oxygen or nitrogen species (ROS/RNS), which are byproducts of cellular oxidative metabolism that carry unstable oxygen radicals¹⁹⁷.
Reactive oxygen/nitrogen species, such as H₂O₂, NO₂•, and superoxides, serve as short-lived paracrine signals that play important physiological roles in immune defense and maintenance of cellular homeostasis¹⁹⁷. Due to the highly reactive property of ROS, the cellular and extracellular levels of ROS are tightly regulated by cellular and enzymatic antioxidants that reduce and stabilize ROS to maintain homeostatic levels of ROS¹⁹⁷. However, an imbalanced and persistent increase of ROS beyond the physiological range depletes the normal antioxidant capacity, resulting in oxidative stress when ROS react with cellular components to cause lipid peroxidation, mitochondrial damage, DNA oxidation, and protein dysfunction¹⁹⁷.
Protein oxidative damage, DNA damage, and cellular death¹⁹⁷. Additionally, subtler changes in cellular ROS levels can alter physiological redox signaling and consequently result in cellular dysfunctions in microglia¹⁹⁸.
In vivo, short-term (2–3 weeks) exposure to concentrated PM or subchronic (3 months) exposure to ambient levels of PM₂.₅ was sufficient to cause oxidative stress in the olfactory bulb and hippocampus of rodents as evidenced by increased lipid peroxidation¹⁵²,¹⁶¹,¹⁹⁹–²⁰¹, dysregulated antioxidant metabolism¹⁴⁹,¹⁵²,²⁰², DNA damage and epigenetic modifications consistent with oxidative stress¹⁰⁷. These findings demonstrate that air pollution can cause oxidative stress that may trigger neuroinflammation via microglia activation.
In tissue culture models, PM₂.₅ or DEP has been shown to increase ROS followed by microglial activation and release of proinflammatory cytokines¹⁵⁷,²⁰³,²⁰⁴, and microglia-endothelial cell coculture²⁰⁵. Interestingly, it has been reported that complement 3 (C3)-mediated microglial activation resulted in aggravated Aβ deposition, synaptic loss, and neuronal degeneration¹⁸²,²⁰⁷–²¹⁰ via NADPH oxidase activation²⁰⁸,²⁰⁹, which increases ROS production. In an independent study, NADPH oxidase activation was found to induce DAM microglial polarization²¹¹.
Exposure to air pollution has also been shown to cause proinflammatory cytokine release in the rodent hippocampus¹⁴⁹,¹⁵⁰,¹⁵²,¹⁵⁸,¹⁶²,¹⁶⁴,²⁰⁰,²¹²–²¹⁴. Across different studies, the main proinflammatory cytokines that were consistently upregulated by PM exposure were TNF-α, IL-1β, IL-6, and IFN-γ. However, whether and how PM-induced proinflammatory cytokines mediate DAM glial polarization and neuronal cell death are still poorly understood.
Several recent in vitro studies suggested potential mechanisms. For example, in a neuron-astrocyte coculture of human cell lines, PM₂.₅ treatment caused the release of chemokines (CCL1, CCL2) that can recruit microglia, as well as cytokines (IL-1β, IFN-γ, IL-5, IL-8) that can polarize microglia into DAM phenotypes¹⁹⁵. Monocultured microglia were polarized to the DAM phenotype (CD86⁺, iNOS⁺ with increased NO release) when exposed to conditioned media from astrocyte–neuron coculture exposed to PM₂.₅; and antibody neutralization of IL-1β, IFN-γ, CCL1, and CCL2 in the conditioned media significantly reduced DAM polarization.
Interestingly, when monocultured microglia were directly exposed to PM₂.₅ they did not polarize to a DAM phenotype, but instead became phagocytic and anti-inflammatory (TREM2⁺, LC3b⁺, CD86⁻), which resulted in effective clearance of PM₂.₅. These authors further demonstrated that microglia and astrocytes both contributed to PM-induced synaptic impairment while DAM microglia primarily drove PM-induced neuronal cell death and tau-phosphorylation¹⁹⁵.
Another in vitro study found that PM₀.₂ induced structural and mitochondrial neuronal atrophy. An independent study further confirmed these results by showing that PM₀.₂-induced neuronal synaptic atrophy was mainly driven via C3 signals released by DAM astrocytes that were polarized by activated microglia. Disease-associated astrocyte polarization was reversed when microglia were inhibited by minocycline even with the PM₀.₂ treatment, and this further reduced synaptic damages¹⁹⁴.
The role of IL-1β has also been investigated in the context of inflammasome activation. The inflammasome is a multiprotein intracellular immune signaling complex that detects pathogen- and damage-associated molecular patterns (PAMPs and DAMPs) to initiate proinflammatory responses and has been repeatedly reported and discussed as a pathogenic mechanism of AD²¹⁵.
In a neuron-microglia coculture, co-exposure to oligomeric Aβ and PM₂.₅ augmented the microglial response to lipopolysaccharide (LPS), evidenced as increased ROS and IL-1β production, resulting in activation of the NLRP3 inflammasome and subsequent neuronal apoptosis.
Coculture with the antioxidant N-acetyl-cysteine and/or caspase-1 inhibitor before oligomeric Aβ and PM₂.₅ exposure significantly ameliorated neuronal apoptosis, suggesting that oxidative stress and NLRP3 inflammasome activation mediate the neurotoxic effects of oligomeric Aβ and PM₂.₅²⁰⁵.
Results from animal studies and in vitro studies corroborate epidemiological findings and provide a better understanding of the molecular and cellular responses to air pollution that include oxidative stress, neuroinflammation, neurodegeneration, and aggravation of AD/ADRD pathologies. While there is ongoing progression towards identifying how air pollutants perturb brain health, mechanistic links that integrate the neuropathological processes triggered by air pollution need further investigation. It is experimentally challenging to recreate realistic air pollution exposure paradigms, but recent technological advances make in vivo simulation of human-relevant exposures to air pollution and ADRD research possible. Nevertheless, discrepancies in exposure paradigms and animal models between studies has led to large variabilities in neurologic outcomes. Therefore, follow-up studies with more standardized exposures using genetically modified animal models with inclusion of both sexes are necessary to validate observations reported across different studies.
Outstanding data gap: Direct vs. indirect effects of air pollution on the brain
While in vitro studies demonstrate that PM from polluted air can directly trigger responses in neurons and glial cells, whether and how gases associated with air pollution directly affect these cell types is largely unknown. Similarly, there remains considerable uncertainty as to whether in vivo brain responses to air pollution arise from direct interactions with air pollutants or rather are the indirect consequence of air pollution effects on peripheral organs. There is experimental evidence, albeit limited, to support both possibilities, which likely are not mutually exclusive.
DIRECT PATHWAY
The direct pathway infers translocation of PM to the brain, where it can directly activate microglia. Translocation of PM to brain parenchyma may occur through retrograde transport of PM from olfactory epithelium to the olfactory cortex via olfactory nerves or via the trigeminal nerve to the brain²¹⁶–²¹⁸ (Figure 3A).
Particulate matter inclusions in the olfactory bulbs and prefrontal cortex have been observed and associated with impaired olfactory function in both mice and humans¹⁰⁹,¹¹⁰,¹¹²,¹¹³,¹⁵⁵,¹⁶⁰,¹⁶¹,¹⁸⁷,²¹⁹,²²⁰.
Alternatively, PM may enter the brain by crossing the blood-brain barrier (BBB) from the systemic circulation. Inhaled fine particles that are deposited in the distal airways and alveoli can readily cross the air–blood interface to enter the systemic circulation and cross vascular barriers like the BBB²²¹–²²⁵. Studies have shown that PM can cross the BBB²²⁵.
Moreover, an in vivo study in which transgenic AD animals were exposed to ambient levels of PM showed extensive PM deposition in the hippocampus (which is vulnerable to BBB leakage in AD), with minimal PM detected in the frontal cortex¹⁶⁰, supporting this as the primary mechanism by which inhaled PM may be reaching the brain under conditions of exposure to ambient TRAP levels.
Blood-brain barrier impairment is a known AD pathology²²⁶–²²⁸ that may be both a causal factor and a result of air-pollution–induced AD exacerbation because PM and circulating mediators can cross an impaired BBB²²⁹ and air pollution can break down components of the neurovascular unit that forms the BBB¹⁹²,¹⁹⁰,²³⁰,²³¹.
Phagocytosis of PM by activated microglia has been well characterized in many in vitro studies¹⁹⁵,²⁰⁵,²⁰⁶,²³²–²³⁴, and numerous materials—metals, endotoxins, and polycyclic aromatic hydrocarbons—adhered to PM have been shown to cause neuroinflammation in the brain parenchyma¹¹⁵,²³⁵–²³⁷. Although there are less
Studies on PM-induced astrocytic activation¹⁶⁹,¹⁹⁴,¹⁹⁵,²⁴³ indicate that the microglia–astrocyte crosstalk can lead to bidirectional responses in both glial cells to promote neuroinflammation¹⁹⁴,¹⁹⁵,²¹⁰,²³⁸,²³⁹.
INDIRECT PATHWAY
The indirect pathway infers that air pollution–associated neuroinflammation is a consequence of inflammation induced by air pollution in peripheral targets. The respiratory toxicology of air pollution has been extensively studied, and it is well-established that inhaled air pollutants reduce lung function and increase respiratory disease susceptibility and mortality by inducing and disrupting pulmonary inflammatory responses²⁴⁰–²⁴³.
Air pollution also causes consequent systemic inflammation, and increased serum levels of proinflammatory cytokines, dysfunctional fibrinolysis, oxidative stress, and activation of circulating immune cells⁴¹,²⁴⁴–²⁴⁸. With growing evidence suggesting that systemic inflammation can promote neuroinflammation²⁴⁹–²⁵⁴ and that higher risks of dementia and cognitive deficits are associated with impaired lung function²⁵⁵–²⁵⁸, the lung–brain axis has been proposed as a potential indirect pathway by which chronic exposure to air pollution induces persistent neuroinflammation that promotes AD/ADRD pathogenesis (Figure 3B).
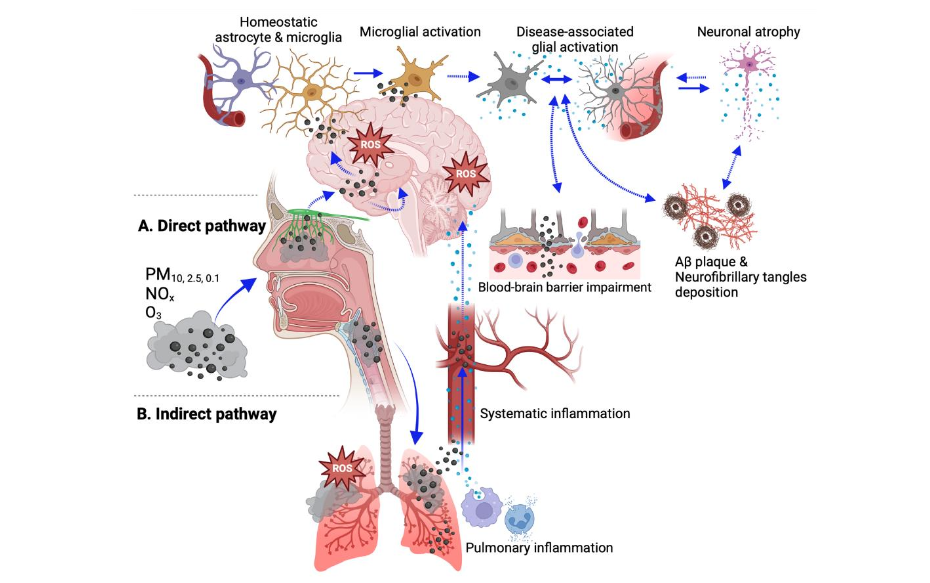
Figure 3. Potential mechanisms by which air pollutants promote neuroinflammation.
Schematic summary of potential pathways by which air pollutants mediate neuroinflammation.
A. Direct pathway:
Inhaled air pollutants infiltrate the brain parenchyma via retrograde transportation from olfactory nerve terminals into the olfactory cortex or by crossing the blood–brain barrier from the systemic circulation to directly activate microglia.B. Indirect pathway:
Inhaled air pollutants cause neuroinflammation via the lung–brain axis. It is hypothesized that air pollutant–induced pulmonary inflammation leads to neuroinflammation via increased systemic inflammation and release of inflammatory mediators into the systemic circulation, which are then delivered to the brain and cross the blood–brain barrier to activate innate immune cells in the brain.NOTE: ROS, reactive oxygen species; dotted arrows indicate hypothesized modes of actions; solid arrows indicate modes of actions supported by experimental evidence.
Created with BioRender.com.While the direct pathway proposes a valid explanation for PM-induced neuroinflammation, it does not adequately explain neuroinflammatory effects of gaseous components like NOₓ and O₃¹⁵⁰. A research group from Indiana University has conducted a series of studies demonstrating how O₃-induced systemic circulating factors regulate microglial and astrocytic activation¹⁵⁰,¹⁵¹,²⁵⁹. An acute exposure to O₃ caused persistent microglial activation in rats 24 hours post-exposure, and the same study showed that the addition of serum from O₃-exposed animals can prime microglia in vitro to augment their response to LPS treatment²⁵⁹.The group’s more recent studies show that O₃ reduced the number of peri-plaque TREM2⁺ microglia but increased peri-plaque colocalization of microglia and astrocytes¹⁵¹ with altered transcriptomic profiles shifting glial cells to more disease-associated states (lower Trem2 and higher Serpina3n gene expressions) in 5xFAD mice¹⁵⁰. They also reported that O₃ increased the expression of damage-associated molecular pattern 1 (HMGB1), which is a DAMP signal that enhances both innate and adaptive immune responses¹⁵⁰. Deleting HMGB1 only in peripheral myeloid cells, but not in microglia by using Hmgb1ᶠˡ/ᶠˡ LysM-Cre⁺ mouse strain, reversed O₃-induced changes in glial expressions and returned Trem2 and Serpina3n transcript levels back to the filtered air control baseline¹⁵⁰,¹⁵¹.An in vitro study from a different group showed that in comparison to directly adding DEP alone to microglia, adding conditioned medium from DEP-stimulated alveolar macrophages caused a higher activation of microglial CD14, which is a pattern recognition receptor that induces DAMP signaling²³⁴. These findings and those from other studies²⁶⁰–²⁶² with similar results suggest that the lung-brain axis may indirectly mediate neuroinflammatory effects of air pollution independent of the direct pathway.
However, further research is required to better understand how the lung-brain axis in different exposure paradigms may not only test acute effects of exclusive air pollutants, but also test long-term effects of exposure to combined air pollutants.
Epigenetics, Environment, and Alzheimer’s disease and related dementias
Epigenetic factors, which mediate gene and environment interactions, may serve as a mechanism underlying G × E interactions and explain a portion of the missing heritability of AD/ADRD⁹³. Epigenetic control is highly complex, with at least 28 known histone, 53 DNA, and 160 RNA modifications, as well as non-coding RNAs, that work in concert to regulate chromatin accessibility, gene expression, mRNA stability, and mRNA translation²⁶³–²⁶⁵.
The location and context-dependent function of these modifications further adds to their complexity but also enables their targeted use as biomarkers and targeted therapeutics²⁶⁶–²⁶⁸. Here, while we do not cover the full breadth of epigenetic modifications and their functions, we do summarize recent findings related to their association with and potential functional roles in G × E interactions between air pollution and AD/ADRD pathogenicity.Prior experimental and population-based epigenetic studies characterizing G × E interactions identified dynamic and disease-associated epigenetic modifications and transcriptional profiles that varied in response to environmental factors²⁶⁸. The modifiable nature of epigenetic modifications and their control over gene expression positions them as prime tools and targets for therapeutic intervention. Additionally, their dynamism makes them well suited to function as biomarkers indicative of disease predisposition, pathological outcomes, and disease susceptibility, or of environmental exposures.
Generally, epigenetic modifications, whether located on histones, DNA, or RNA, are regulated by writer and eraser enzymes and interpreted by reader proteins or protein/RNA complexes. The expression of these regulatory factors enables fine control over tissue- and cell-type–specific spatiotemporal expressional programming²⁶⁹–²⁷². Dysregulation of epigenetic control, either through he alteration of specific markers or the abundance of the readers/writers/erasers serves as an additional mechanism of control and potential contributor towards disease state.
DNA MODIFICATIONS
Recent work characterizing the relative contributions of genetic and environmental factors in the development and progression of AD highlighted epigenetic modifications as critical mediators of G × E interactions. In a cohort of older women from the Women’s Health Initiative Memory Study (WHIMS), it was found that nanoparticles exposure increased the risk of cognitive decline by 81% and all-cause dementia by 92% in a dose-dependent manner for APOE ε4 carriers²⁷³.
Following up on these findings, the authors utilized the 5xFAD mouse model and found that exposure to nanoparticles was associated with greater AD pathology and the APOE status of the exposed individual functioned as a response modifier. In the 5xFAD mouse model, even low doses of 40–60 nm nanoparticles exposure, the presence of ApoE ε4/+ over ApoE ε3/+ alleles significantly increased amyloid plaque pathology, reduced hippocampal CA1 neurites, and decreased the glutamate GluR1 subunit.In a similar study investigating the G × E interaction between cadmium exposure and ApoE ε4 dosage, 14 weeks of cadmium exposure in the ApoE knock-in mouse model identified accelerated cognitive impairment and reduced hippocampal neurogenesis in ApoE ε4 carriers relative to their ApoE ε3 counterparts, indicating G × E interactions modulate AD pathogenicity²⁷⁴.In an effort to explore the missing heritability observed in AD, Panitch et al. recently investigated the relationship between APOE DNA methylation (5-methylcytosine; 5mC) and ε4 carrier status and found differential methylation at 25 CpG sites in the dorsolateral prefrontal cortex and 36 CpG sites in blood, with the majority of sites being hypomethylated²⁷⁵. Furthermore, this group identified seven CpG sites in the APOE region (including TOMM40, APOE, and APOC1 genes) that significantly differed between APOE ε4 carriers and non-carriers in brain and blood (P < 5 × 10⁻⁸), with three sites in the APOE gene showing hypermethylation in ε4 carriers and a nominal association with APOE expression in the brain (P < 10⁻⁵)²⁷⁵.One of the three genes that contained differentially methylated CpG sites in APOE ε4 carriers relative to control was TET1, a 5mC eraser and DNA hydroxymethylation (5-hydroxymethylcytosine, 5hmC) writer. In the same family of Ten-Eleven Translocation proteins, TET2, a 5mC eraser and 5hmC writer, was also found to be moderately differentially methylated in APOE ε4 carriers relative to control (p = 7.6 × 10⁻⁷)²⁷⁵.
Interestingly, both TET1 and TET2 function in similar but distinct capacities, and prior sequencing studies have identified an enrichment of TET1 and TET2 variants associated with EOAD/frontotemporal dementia²⁷⁶,²⁷⁷. Lastly, utilizing an Aβ mouse model, the authors found that the dioxygenase 5mC eraser/writer Dnmt3a, to dCas9 targeted to APP was associated with the Amyloid Precursor Protein pathway and increased cognitive and behavioral impairment²⁷⁹.
HISTONE MODIFICATIONS
In addition to DNA modifications, histone modifications have emerged as crucial players in the pathogenesis of AD and often function with DNA modifications to regulate gene expression²⁷⁹,²⁸⁰.
Recent studies reveal links between air pollution and histone modifications. Zheng et al. explored how different components of air pollution (PM₂.₅, PM₁₀, black carbon, and elemental components—potassium, sulfur, iron, silicon, aluminum, zinc, calcium, and titanium) influenced various histone modifications (H3K9ac, H3K9me3, H3K27me3, and H3K36me3) in the blood leukocytes of exposed truck drivers and office workers in Beijing²⁸¹. Their study identified differential associations between pollutants and various histone markers.Specifically, they noted an increase in ambient PM₂.₅ associated with lower H3K27me3 and H3K36me3 levels. They also observed that office workers had a stronger association between black carbon and H3K9ac and H3K36me3 than truck driverscarbon exposure and H3K9ac and H3K9me3 status was sex-dependent²⁸¹.
Similarly, exposure to air pollutants, particularly PM₂.₅ and PM₁₀, induced significant epigenetic alterations in rat blood and lung tissue in a dose-dependent manner²⁸². This study revealed that increased exposure to PM₂.₅ and PM₁₀ generally led to decreased DNA methylation of LINE-1 and iNOS, while simultaneously increasing histone acetylation (H3K9ac) in both blood and lung tissue. Interestingly, PM₂.₅ exposure was also associated with increased methylation of p16CDKN2A and APC promoters. The effects of NOₓ were more variable, showing mixed impacts on methylation patterns. Notably, these epigenetic changes were often more pronounced in blood compared to lung tissue, with H3K9ac consistently increasing in response to PM₂.₅ and PM₁₀ exposure in both tissue types²⁸².
RNA MODIFICATIONS
RNA N6-methyladenosine (m⁶A) is the most abundant internal modification in eukaryotic mRNAs, highly enriched in the brain, and associated with a suite of neurodevelopmental and neurodegenerative diseases, including AD. m⁶A influences transcript splicing, stability, translation, nuclear export, and RNA structure²⁸³,²⁸⁴.
There is evidence that environmental exposure influences RNA modifications and subsequently AD pathophysiology²⁸⁵. Li et al., 2023 found that exposure to PM₂.₅ is associated with a greater prevalence of m⁶A modifications, a global increase in gene expression, and a significant increase in the expression of prostaglandin-endoperoxide synthase 2 (PTGS2), which is involved in synthesizing the prostaglandin inflammation signaling molecules²⁸⁶.Recent research shed light on the molecular mechanisms underlying the effects of PM₂.₅ exposure on lung cells²⁸⁷. In the A549 lung cell line, PM₂.₅ exposure upregulated m⁶A RNA methylation coincident with increased expression of TGF-β, SMAD3, and the methyltransferases METTL3 and METTL14 (m⁶A writers). Importantly, inhibition of TGF-β reversed the PM₂.₅-induced changes, suggesting a pivotal role for the TGF-β signaling pathway in regulating m⁶A RNA methylation following PM₂.₅ exposure²⁸⁷. These findings revealed a potential mechanism by which lung inflammation triggered by PM₂.₅ exposure leads to m⁶A modifications observed in AD.Using postmortem AD patient data, another study identified significantly reduced expression and soluble protein levels of METTL3, and no significant changes for METTL14 in the hippocampus²⁸⁸. Notably, insoluble fractions of AD brain samples had accumulated METTL3 that positively correlated with insoluble tau protein levels, although no direct interaction between METTL3 and tau was observed²⁸⁸. This research suggests that while tau pathology is a better predictor of disrupted m⁶A signaling than Aβ load, it likely does not directly cause the altered METTL3 expression seen in AD hippocampal neurons.These findings were corroborated in a study of individuals diagnosed with AD vs. controls that reported increased m⁶A modifications in cortex and hippocampus tissue with AD. The functional significance of this observation is suggested by experimental animal studies: knockdown of METTL3 in mouse hippocampus significantly increased memory loss, neurodegeneration, gliosis, oxidative stress, and apoptotic processes²⁸⁹. Knocking down METTL3, METTL14, and YTHDF in flies expressing TauR406W was found to worsen eye phenotype and reduce motor function, highlighting that reduced expression of m⁶A or the ability to read m⁶A is associated with worsened AD-associated phenotypes²⁹⁰.
Collectively, these studies suggest that the regulation of RNA writers, readers and erasers is a critical component for mediating G × E interactions, and aberrant regulation of these proteins has a large knock-on effect, influencing pathological outcomes.
NON-CODING RNA
A separate class of RNAs, non-coding RNAs, exert epigenetic control via control of transcription, transcript stability Molecules are highly dynamic, and their expression and stability can be influenced by exposure to environmental factors²⁹¹–²⁹⁴.To determine whether DEP induces pulmonary inflammatory response, Wang and colleagues explored the involvement of RNA binding protein LIN28B and non-coding microRNA let7 axis in the inflammatory response to DEP exposure in mice²⁹⁵. Let7-d and let7-g were significantly downregulated, while LIN28B protein levels were elevated from 6 to 24 hours after DEP exposure. Furthermore, they found that LIN28B is responsible for the downregulation of let7, subsequently increasing expression of the pro-inflammatory cytokine IL-6²⁹⁵.A study involving a protein of the same class, LIN28A, in a VaD rat model, where VaD was introduced by bilateral common carotid artery occlusion, found that treatment with Lin28a siRNA ameliorated cognitive impairment, as well as upregulated expression of GFAP, and IBA-1 expression markers²⁹⁶. Moreover, treatment with siRNA alleviated BBB damage as measured by expression of PECAM-1, PDGFRβ, occludin, claudin-9, and ZO-1, and Ccr6²⁹⁶.Furthering the link between environmental exposure, non-coding RNAs and modulation of inflammation, microarray analyses of blood samples from foundry workers exposed to metal-rich PM identified significantly increased expression of miR-421, miR-146a, miR-29a, and let-7g after 3 days of work exposure relative to baseline (first day of the work week after two days off)²⁹⁷. In addition to sampling miRNA expression, PCR was performed on 18 inflammatory genes including TGFB1, TGFB2, TNF, ITGA2, ITGAX, NFKB1, NOS2, CCL2, CCL5. Significant differences were found in individual miRNAs and inflammatory genes that have differing targets and regulatory activity. This included miR-421 associated with NOS2 expression; miR-146a with TGFB1, CCL2, and CCL5 expression; and let-7g with TGFB1, TGFB2, and NFKB1 expression²⁹⁷.Additional research exploring the relationship between PM₂.₅ exposure and expression of non-coding RNAs has identified increased expression of the long non-coding RNA HCG18 in the lung. HCG18 suppresses miR-195 expression and leads to upregulated ATG14 expression and increased autophagy and progression of lung adenocarcinoma²⁹⁸. In addition to playing a significant role in the development of lung pathologies, reduced miR-195 expression is observed with human aging and in AD progression, while increased miR-195 prevalence is associated with improved cognitive performance and negatively correlated with cerebrospinal fluid tau levels. Human expression patterns of miR-195 are mirrored in ApoE ε4/+ mice when compared with ApoE ε3/+ mice. Furthermore, elevating miR-195 levels was found to reduce amyloid burden and tau hyperphosphorylation and improve cognitive impairment. Lastly, when cortical neurons and astrocytes generated from inducible pluripotent stem cells derived from ApoE ε4/+ mice were co-cultured, miR-195 inhibition exacerbated AD-associated pathogenic phenotypes²⁹⁹.In this section, we highlighted epigenetic mechanisms by which air pollutants may contribute to cognitive impairment and AD-associated pathologies. These studies underscore the crucial role of epigenetics as a mediator of G × E interactions in AD. Importantly, they indicate that the development of epigenetic markers of air pollution exposure may advance the identification of specific air pollutants that promote AD/ADRD-associated risk and progression. Epigenetic analyses also hold the promise of providing targets for therapeutic interventions, offering modifiable pathways to potentially reduce AD pathophysiology.
Conclusions
Consistent with previous epidemiological findings regarding the association between air pollution and ADRD, recent epidemiological studies have largely reported positive correlations between air pollution and AD/ADRD. This association is mostly substantiated by experimental animal studies. Although there is general consensus that air AD/ADRD, associations between specific air pollutants and ADRD subtypes are variable across different studies. This variability in part stems from the complexity of mixed ADRD pathologies and equivocal etiological links between genetic status, pathological manifestation, cognitive impairment, and clinical diagnoses. Currently, the field is reassessing diagnostic criteria utilizing biomarkers that more accurately delineate between AD and related dementias to better characterize pathology-specific disease progression. These refinements in diagnostic precision will improve efforts to identify environmental factors that modify risk and severity of AD/ADRD. Elucidating ADRD stratification and G × E contributions implicated in the development of ADRD will inform effective public health regulations and guidelines for air pollution and other environmental contaminants, with the aim of preventing and minimizing adverse neurological health outcomes.
Conflict of Interest:
The authors declare that they have no conflicts of interest.
Funding Statement:
This work was supported by the National Institute on Aging (grant # RF1 AG074709 to PJL), the National Institute of Neurological Disorders and Stroke (grant # RF1 NS130659 to PJL) and the National Heart, Lung, and Blood Institute predoctoral training grant program (grant # T32 HL007013 to HHP).
Acknowledgements:
The authors thank Dr. Suzette Smiley-Jewell (UC Davis) for her assistance in proofreading and editing this manuscript.
List of abbreviations
| Abbreviation | Definition |
|---|---|
| 5hmC | 5-hydroxymethylcytosine |
| 5mC | 5-methylcytosine |
| AD | Alzheimer’s disease |
| ADRD | Alzheimer’s disease-related dementias |
| Aβ | Amyloid beta plaques |
| BBB | Blood-brain barrier |
| BC | Black carbon |
| C3 | Complement 3 |
| CAP | Concentrated ambient particle |
| CO | Carbon monoxide |
| CSF | Cerebrospinal fluid |
| DAM | Disease-associated molecular biomarkers |
| DAMP | Damage-associated molecular pattern |
| DEP | Diesel exhaust particles |
| EOAD | Early-onset Alzheimer’s disease |
| G × E | Gene by environment interaction |
| HMGB1 | High mobility group box 1 |
| LOAD | Late-onset Alzheimer’s disease |
| LPS | Lipopolysaccharide |
| m⁶A | N6-methyladenosine |
| MRI | Magnetic resonance imaging |
| Abbreviation | Definition |
|---|---|
| NFL | Neurofilament light |
| NFT | Neurofibrillary tau tangles |
| NOₓ | Nitrogen oxides |
| O₃ | Ground-level ozone |
| PAMP | Pathogen-associated molecular pattern |
| PET | Positron emission tomography |
| PFAS | Per- and polyfluoroalkyl substances |
| PM | Particulate matter |
| PTGS2 | Prostaglandin-endoperoxide synthase 2 |
| ROS/RNS | Reactive oxygen or nitrogen species |
| SO₂ | Sulfur dioxide |
| TRAP | Traffic-related air pollution |
| UFPM | Ultrafine particulate matter |
| VaD | Vascular dementia |
References
- Collaborators GBDDF. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: an analysis for the Global Burden of Disease Study 2019. Lancet Public Health. Feb 2022;7(2):e105-e125. doi:10.1016/S2468-2667(21)00249-8
- Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. Jun 2016;8(6):595-608. doi:10.15252/emmm.201606210
- Jack CR, Jr., Bennett DA, Blennow K, et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. Apr 2018;14(4):535-562. doi:10.1016/j.jalz.2018.02.018
- Khan SS, Bloom GS. Tau: The Center of the Signaling Nexus in Alzheimer’s Disease. Front Neurosci. 2016;10:31. doi:10.3389/fnins.2016.00031
- 2024 Alzheimer’s disease facts and figures. Alzheimers Dement. May 2024;20(5):3708-3821. doi:10.1002/alz.13809
- Dubois B, Hampel H, Feldman HH, et al. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimers Dement. Mar 2016;12(3):292-323. doi:10.1016/j.jalz.2016.02.002
- Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. May 2011;7(3):280-92. doi:10.1016/j.jalz.2011.03.003
- Petersen RC, Caracciolo B, Brayne C, Gauthier S, Jelic V, Fratiglioni L. Mild cognitive impairment: a concept in evolution. J Intern Med. Mar 2014;275(3):214-28. doi:10.1111/joim.12190
- Petersen RC, Lopez O, Armstrong MJ, et al. Practice guideline update summary: Mild cognitive impairment: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology. Neurology. Jan 16 2018;90(3):126-135. doi:10.1212/WNL.0000000000004826
- Dunn AR, O’Connell KMS, Kaczorowski CC. Gene-by-environment interactions in Alzheimer’s disease and Parkinson’s disease. Neurosci Biobehav Rev. Aug 2019;103:73-80. doi:10.1016/j.neubiorev.2019.06.018
- Eid A, Mhatre I, Richardson JR. Gene-environment interactions in Alzheimer’s disease: A potential path to precision medicine. Pharmacol Ther. Jul 2019;199:173-187. doi:10.1016/j.pharmthera.2019.03.005
- Livingston G, Huntley J, Sommerlad A, et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet. Aug 8 2020;396(10248):413-446. doi:10.1016/S0140-6736(20)30367-6
- Shindler EE, Galasko D, Perrin R, et al. Acceptable performance of blood biomarkers in global amyloid pathology – recommendations from the Blood Biomarker Working Group. Alz Res Neurol. Jul 2024;20(7):426-439. doi:10.1038/s41582-024-00977-5
- Ritchie C, Smailagic N, Noel-Storr AH, Ukoumunne O, Laddis EC, Martin S. CSF tau and the CSF tau/Aβeta ratio for the diagnosis of Alzheimer’s disease dementia and other dementias in people with mild cognitive impairment (MCI). Cochrane Database Syst Rev. Mar 22 2017;3(3):CD010803. doi:10.1002/14651858.CD010803.pub2
- National Plan to Address Alzheimer’s Disease: 2023 Update. Updated 2024, April 22. https://aspe.hhs.gov/reports/national-plan-2023-update
- Schneider JA. Neuropathology of Dementia Disorders. Continuum (Minneap Minn). Jun 1 2022;28(3):834-851. doi:10.1212/CON.0000000000001137
- Arvanitakis Z, Shah RC, Bennett DA. Diagnosis and Management of Dementia Review. JAMA. Oct 23 2019;322(16):1589-1599. doi:10.1001/jama.2019.4782
- Caldwell AB, Anantharaman BG, Ramadhason S, et al. Transcriptomic profiling of Alzheimer’s disease patients. Mol Brain. Oct 12 2022;15(1):83. doi:10.1186/s13041-022-00963-2
- 19. Reitz C, Rogaeva E, Beecham GW. Late-onset vs nonmendelian early-onset Alzheimer disease: A distinction without a difference? Neurol Genet. Oct 2020;6(5):e512. doi:10.1212/NXG.0000000000000512
- 20. Dao L, Choi S, Freeby M. Type 2 diabetes mellitus and cognitive function: understanding the connections. Curr Opin Endocrinol Diabetes Obes. Feb 1 2023;30(1):7-13. doi:10.1097/MED.0000000000000783
- 21. Benedict RHB, Amato MP, DeLuca J, Geurts JJG. Cognitive impairment in multiple sclerosis: clinical management, MRI, and therapeutic avenues. Lancet Neurol. Oct 2020;19(10):860-871. doi:10.1016/S1474-4422(20)30277-5
- 22. McInnes K, Friesen CL, MacKenzie DE, Westwood DA, Boe SG. Mild Traumatic Brain Injury (mTBI) and chronic cognitive impairment: A scoping review. PLoS One. 2017;12(4):e0174847. doi:10.1371/journal.pone.0174847
- 23. Robinson JL, Richardson H, Xie SX, et al. The development and convergence of co-pathologies in Alzheimer’s disease. Brain. Apr 12 2021;144(3):953-962. doi:10.1093/brain/awaa438
- 24. Greenberg SM, Backsai BJ, Hernandez-Guillamon M, Pruzin J, Sperling R, van Veluw SJ. Cerebral amyloid angiopathy and Alzheimer disease – one peptide, two pathways. Nat Rev Neurol. Jan 2020;16(1):30-42. doi:10.1038/s41582-019-0281-2
- 25. Grasso D, Castorani G, Borreggine C, Simeone A, De Blasi R. Cerebral amyloid angiopathy related inflammation: A little known but not to be underestimated disease. Radiol Case Rep. Sep 2021;16(9):2514-2521. doi:10.1016/j.radcr.2021.05.080
- 26. Hereditary cerebral amyloid angiopathy (2022).
- 27. Carmona EM, Tomassein J, Ossenkoppele R, et al. Genetically identical twins show comparable tau PET load and spatial distribution. Brain. Oct 21 2022;145(10):3573-3581. doi:10.1093/brain/awac202
- 28. Puckett OK, Fennema-Notestine C, Hagler DJ, Jr, et al. “The Association between Exposure to Fine Particulate Matter and MRI-Assessed Locus Coeruleus Integrity in the Vietnam Era Twin Study of Aging (VETSA). Environ Health Perspect. Jul 2024;132(7):77006. doi:10.1289/EHP14344
- 29. Karlsson IK, Escott-Price V, Gatz M, et al. Measuring heritable contributions to Alzheimer’s disease: polygenic risk score analysis with twins. Brain Commun. 2022;4(1):fcab308. doi:10.1093/braincomms/fcab308
- 30. Varjonen A, Schwarz C, Vuoksimaa E. Co-twin design in brain imaging—review on biomarkers of Alzheimer’s disease. Cereb Cortex. Jul 5 2023;33(14):9054-9066. doi:10.1093/cercor/bhad181
- 31. Livingston G, Sommerlad A, Orgeta V, et al. Dementia prevention, intervention, and care. Lancet. Dec 16 2017;390(10113):2673-2734. doi:10.1016/S0140-6736(17)31363-6
- 32. Gunnarsson LG, Bodin L. Occupational Exposures and Neurodegenerative Disease–A Systematic Literature Review and Meta-Analyses. Int J Environ Res Public Health. Mar 12 2020;17(6):2337. doi:10.3390/ijerph17062337
- 33. Sarailoo M, Afshari S, Asghariazar V, Safarzadeh E, Dadkhah M. Cognitive Impairment and Neurodegenerative Diseases Development Associated with Organophosphate Pesticides Exposure: a Review Study. Neurotox Res. Oct 2022;40(5):1624-1643. doi:10.1007/s12640-022-00552-0
- 34. Kim JY, Park SJ, Kim SK, et al. Pesticide exposure and cognitive decline in a rural South Korean population. PLoS One. 2019;14(3):e0213738. doi:10.1371/journal.pone.0213738
- 35. Hayden KM, Norton MC, Darcey D, et al. Occupational exposure to pesticides increases the risk of incident AD: the Cache County study. Neurology. May 11 2010;74(19):1524-30. doi:10.1212/WNL.0b013e3181dd4423
- 36. Sturm ET, Castro C, Mendez-Colmenares A, et al. Risk Factors for Brain Health in Agricultural Work: A Systematic Review. Int J Environ Res Public Health. Mar 12 2020;17(6):2337. doi:10.3390/ijerph17063337
- 37. Pales VA, Suski KG, Baker JM, et al. Manganese exposure and working memory- related brain activity in smallholder farmworkers in Costa Rica: Results from a pilot study. Environ Res. Jun 2019;173:539-548. doi:10.1016/j.envres.2019.04.006
- 38. Liu CH, Huang CY, Huang CC. Occupational neurotoxic diseases in taiwan. Saf Health Work. Dec 2012;3(4):257-67. doi:10.5491/SHAW.2012.3.4.257
- 39. Baldi I, Lebailly P, Mohammed-Brahim B, Letenneur L, Dartigues JF, Brochard P. Neurodegenerative diseases and exposure to pesticides in the elderly. Am J Epidemiol. Mar 1 2003;157(5):409-14. doi:10.1093/aje/kwf216
- 40. Delcourt N, Pouget AM, Grivaud A, et al. First Observations of a Potential Association Between Accumulation of Per- and Polyfluoroalkyl Substances in the Central Nervous System and Markers of Alzheimer’s Disease. J Gerontol A Biol Sci Med Sci. Mar 1 2024;79(3) doi:10.1093/gerona/glad024
- 41. Lederer AM, Fredriksen PM, Nkeh-Chungag BN, et al. Air pollution and brain aging: current evidence from animal and human studies. Am J Physiol Heart Circ Physiol. Apr 1 2021;320(4):H1417-H1439. doi:10.1152/ajpheart.00706.2020
- 42. Anderson JO, Thundiyil JG, Stolbach A. Clearing the air: a review of the effects of particulate matter air pollution on human health. J Med Toxicol. Jun 2012;8(2):166-75. doi:10.1007/s13181-011-0203-1
- 43. Bennett M, Nault I, Koehle M, Wilton S. Air Pollution and Arrhythmias. Can J Cardiol. Sep 2023;39(9):1253-1262. doi:10.1016/j.cjca.2023.03.023
- 44. Brook RD, Rajagopalan S, Pope CA, 3rd, et al. Particulate matter air pollution and cardiovascular disease: An update to the scientific statement from the American Heart Association. Circulation. Jun 1 2010;121(21):2331-78. doi:10.1161/CIR.0b013e3181dbbec1
- 45. Fiordelisi A, Piscitelli P, Trimarco B, Coscioni E, Iaccarino G, Sorriento D. The mechanisms of air pollution and particulate matter in cardiovascular diseases. Heart Fail Rev. May 2017;22(3):337-347. doi:10.1007/s10741-017-9606-7
- 46. Miller MR, Newby DE. Air pollution and cardiovascular disease: car sick. Cardiovasc Res. Feb 1 2020;116(2):279-294. doi:10.1093/cvr/cvz228
- 47. Rajagopalan S, Brook RD, Salerno P, et al. Air pollution exposure and cardiometabolic risk. Lancet Diabetes Endocrinol. Mar 2024;12(3):196-208. doi:10.1016/S2213-8587(23)00361-3
- 48. Tainio M, Jovanovic Andersen Z, Nieuwenhuijsen MJ, et al. Air pollution, physical activity and health: A mapping review of the evidence. Environ Int. Feb 2021;147:105954. doi:10.1016/j.envint.2020.105954
- 49. Paul KC, Haan M, Mayeda ER, Ritz BR. Ambient Air Pollution, Noise, and Late-Life Cognitive Decline and Dementia Risk. Annu Rev Public Health. Apr 1 2019;40:203-220. doi:10.1146/annurev-publhealth-040218-044058
- 50. Mok B, Brun D, Zanobetti A. Time-lagged relationships between air pollution and Alzheimer’s mortality in the United States: A county-level study. Environ Int. Dec 2022;163:107694. doi:10.1016/j.envint.2022.107694
- 51. Zhang B, Weuve J, Langa KM, et al. Comparison of Particulate Air Pollution From Different Emission Sources and Incident Dementia in the US. JAMA Intern Med. Aug 14 2023; doi:10.1001/jamainternmed.2023.3300
- 52. Mortamais M, Gutierrez LA, de Hoogh K, et al. Long-term exposure to ambient air pollution and risk of dementia: Results of the prospective Three-City Study. Environ Int. Mar 2021;148:106376. doi:10.1016/j.envint.2020.106376
- 53. Iaccarino L, La Joie R, Lesman-Segev OH, et al. Association Between Ambient Air Pollution and Amyloid Positron Emission Tomography Positivity in Older Adults With Cognitive Impairment. JAMA Neurol. Feb 1 2021;78(2):197-207. doi:10.1001/jamaneurol.2020.3962
- 54. Hajat A, Park C, Adam C, et al. Air pollution and plasma amyloid beta in a cohort of older adults: Evidence from the Ginkgo Evaluation of Memory Study. Environ Int. Feb 2023;172:107080. doi:10.1016/j.envint.2023.107080 55. Weuve J, Puett RC, Schwartz J, Yanosky JD, Laden F, Grodstein F. Exposure to particulate air pollution and cognitive decline in older women. Arch Intern Med. Feb 13 2012;172(3):219-27. doi:10.1001/archinternmed.2011.683
- 56. Ranft U, Schikowski T, Sugiri D, Krutmann J, Kramer U. Long-term exposure to traffic-related particulate matter impairs cognitive function in the elderly. Environ Res. Nov 2009;109(8):1004-11. doi:10.1016/j.envres.2009.08.003
- 57. Jung CR, Lin YT, Hwang BF. Ozone, particulate matter, and newly diagnosed Alzheimer’s disease: a population-based cohort study in Taiwan. J Alzheimers Dis. 2015;44(2):573-84. doi:10.3233/JAD-140855
- 58. Shi L, Steenland K, Li H, et al. A national cohort study (2000–2018) of long-term air pollution exposure and incident dementia in older adults in the United States. Nat Commun. Nov 19 2022;12(1):6754. doi:10.1038/s41467-021-27049-2
- 59. Yuan S, Huang X, Zhang L, et al. Associations of air pollution with all-cause dementia, Alzheimer’s disease, and vascular dementia: a prospective cohort study based on 437,932 participants from the UK biobank. Front Neurosci. 2023;17:1216686. doi:10.3389/fnins.2023.1216686
- 60. Oudin A, Segersson D, Adolfsson R, Forsberg B. Association between air pollution from residential wood burning and dementia incidence in a longitudinal study in Northern Sweden. PLoS One. 2018;13(6):e0198283. doi:10.1371/journal.pone.0198283
- 61. Carey IM, Anderson HR, Atkinson RW, et al. Are noise and air pollution related to the incidence of dementia? A cohort study in London, England. BMJ Open. Sep 11 2018;8(9):e022404. doi:10.1136/bmjopen-2018-022404
- 62. Parra KL, Alexander GE, Raichlen DA, Klimentidis YC, Furlong MA. Exposure to air pollution and risk of incident dementia in the UK Biobank. Environ Res. Jun 2022;209:112895. doi:10.1016/j.envres.2022.112895
- 63. Zhang H, Shi L, Ebelt ST, et al. Short-term associations between ambient air pollution and emergency department visits for Alzheimer’s disease and related dementias. Environ Epidemiol. Feb 2023;7(1):e237. doi:10.1097/EE9.0000000000000237
- 64. Rhew SH, Kravchenko J, Lyerly HK. Exposure to low-dose ambient fine particulate matter PM2.5 and Alzheimer’s disease, non-Alzheimer’s dementia, and Parkinson’s disease in North Carolina. PLoS One. 2021;16(7):e0253253. doi:10.1371/journal.pone.0253253
- 65. Wu J, Jackson L. Greenspace Inversely Associated with the Risk of Alzheimer’s Disease in the Mid-Atlantic United States. Earth (Basel). Feb 28 2021;2(1):140-150. doi:10.3390/earth2010009
- 66. Zhang Z, Chen L, Wang X, et al. Associations of Air Pollution and Genetic Risk with Incident Alzheimer’s Disease: A Prospective Cohort Study. Am J Epidemiol. Feb 1 2023;192(2):182-194. doi:10.1093/aje/kwac188
- 67. Bellander T, Rizzuto D. Association Between Cardiovascular Disease and Long-term Exposure to Air Pollution With the Risk of Dementia. JAMA Neurol. Jul 1 2020;77(7):801-809. doi:10.1001/jamaneurol.2019.4914
- 68. Grande G, Hooshmand B, Vetrano DL, et al. Association of Long-term Exposure to Air Pollution and Dementia Risk: The Role of Homocysteine, Methionine, and Cardiovascular Burden. Neurology. Jul 13 2023; doi:10.1212/WNL.0000000000207656
- 69. Ilango SD, Chen H, Hystad P, et al. The role of cardiovascular disease in the relationship between air pollution and incident dementia: a population-based cohort study. Int J Epidemiol. Feb 1 2020;49(1):36-44. doi:10.1093/ije/dyz154
- 70. Ilango SD, Leary CS, Ritchie E, et al. An Examination of the Joint Effect of the Social Environment and Air Pollution on Dementia Among US Older Adults. Environ Epidemiol. Jun 2023;7(3):e250. doi:10.1097/EE9.0000000000000250
- 71. Liu T, Zhou Y, Wei J, et al. Association between short-term exposure to ambient air pollution and dementia mortality in Chinese adults. Sci Total Environ. Nov 25 2022;849:157860. doi:10.1016/j.scitotenv.2022.157860
- 72. Ma H, Li X, Zhou T, Wang M, Heianza Y, Qi L. Long-term exposure to low-level air pollution, genetic susceptibility and risk of dementia. Int J Epidemiol. Jun 6 2023;52(3):738-748. doi:10.1093/ije/dyac146
- 73. Oudin A, Andersson J, Sundstrom A, et al. Traffic-Related Air Pollution as a Risk Factor for Dementia: No Clear Modifying Effects of APOEvarepsilon4 in the Betula Cohort. J Alzheimers Dis. 2019;71(3):733-740. doi:10.3233/JAD-181037
- 74. Shi L, Zhu Q, Wang Y, et al. Incident dementia and long-term exposure to constituents of fine particle air pollution: A national cohort study in the United States. Proc Natl Acad Sci U S A. Jan 3 2023;120(1):e2211282119. doi:10.1073/pnas.2211282119
- 75. Smargiassi A, Sidi EAL, Robert E, et al. Exposure to ambient air pollutants and the onset of dementia in Quebec, Canada. Environ Res. Nov 2020;190:109870. doi:10.1016/j.envres.2020.109870
- 76. So R, Andersen ZJ, Chen J, et al. Long-term exposure to air pollution and mortality in a Danish nationwide administrative cohort study: Beyond mortality from cardiopulmonary disease and lung cancer. Environ Int. Jun 2022;164:107241. doi:10.1016/j.envint.2022.107241
- 77. Wang X, Younan D, Millstein J, et al. Association of improved air quality with lower dementia risk in older women. Proc Natl Acad Sci USA. Jan 11 2022;119(2):e*. doi:10.1073/pnas.2107833119
- 78. Yu Y, Su J, Jerrett M, et al. Air pollution and traffic noise interact to affect cognitive health in older Mexican Americans. Environ Int. Mar 2023;173:107810. doi:10.1016/j.envint.2023.107810
- 79. Yuchi W, Sbihi H, Davies H, Tamburic L, Brauer M. Road proximity, air pollution, noise, green space and neurologic disease incidence: a population-based cohort study. Environ Health. Jan 21 2020;19(1):8. doi:10.1186/s12940-020-00565-4
- 80. Zhu Z, Yang Z, Yu L, et al. Residential greenness, air pollution and incident neurodegenerative disease: A cohort study in China. Sci Total Environ. Jun 20 2023;878:163173. doi:10.1016/j.scitotenv.2023.163173
- 81. Tzivian L, Dlugaj M, Winkler A, et al. Long-Term Air Pollution and Traffic Noise Exposures and Mild Cognitive Impairment in Older Adults: A Cross-Sectional Analysis of the Heinz Nixdorf Recall Study. Environ Health Perspect. Sep 2016;124(9):1361-8. doi:10.1289/ehp.1509824
- 82. Cerza F, Renzi M, Gariazzo C, et al. Long-term exposure to air pollution and hospitalization for dementia in the Rome longitudinal study. Environ Health. Aug 9 2019;18(1):72. doi:10.1186/s12940-019-0511-5
- 83. Ge R, Wang Y, Zhang Z, Sun H, Chang J. Association of long-term exposure to various ambient air pollutants, lifestyle, and genetic factors with dementia: A prospective cohort study. Environ Health. Jan 25 2024;24(1):179. doi:10.1186/s12889-024-17702-y
- 84. Petters S, Bouma F, Heck G, Janssen N, Vermeulen R. Air pollution exposure and mortality from neurodegenerative diseases in the Netherlands: A population-based cohort study. Environ Res. Jul 2 2024;259:119552. doi:10.1016/j.envres.2024.119552
- 85. Tian F, Qian Z, Zhang Z, et al. Air pollution, APOE genotype and risk of dementia among individuals with cardiovascular diseases: A population-based longitudinal study. Environ Pollut. Apr 15 2024;347:123758. doi:10.1016/j.envpol.2024.123758
- 86. Wang J, Hu X, Yang T, et al. Ambient air pollution and the dynamic transitions of stroke and dementia: a population-based cohort study. ELClinicalMedicine. Jan 2024;67:102368. doi:10.1016/j.eclinm.2023.102368
- 87. Sturza J, Zhang J, Bergmann M, et al. Long-term exposure to air pollution and road traffic noise and incidence of dementia in the Danish Nurse Cohort. Alzheimers Dement. Jun 2024;20(6):4080-4091. doi:10.1002/alz.13814 88. Zhang B, Langa KM, Weuve J, et al. Hypertension and Stroke as Mediators of Air Pollution Exposure and Incident Dementia. JAMA Netw Open. Sep 5 2023;6(9):e2333470. doi:10.1001/jamanetworkopen.2023.33470
- 89. Shaffer RM, Blanco MN, Li G, et al. Fine Particulate Matter and Dementia Incidence in the Adult Changes in Thought Study. Environ Health Perspect. Aug 2021;129(8):087001. doi:10.1289/EHP9018
- 90. Wu QZ, Zeng HX, Andersson J, et al. Long-term exposure to major constituents of fine particulate matter and neurodegenerative diseases: A population-based survey in the Pearl River Delta Region, China. J Hazard Mater. May 15 2024;470:134161. doi:10.1016/j.jhazmat.2024.134161
- 91. Paul KC, Haan M, Yu Y, et al. Traffic-Related Air Pollution and Incident Dementia: Direct and Indirect Pathways Through Metabolic Dysfunction. J Alzheimers Dis. 2020;76(4):1477-1491. doi:10.3233/JAD-200232
- 92. Andersson J, Oudin A, Sundstrom A, Forsberg B, Adolfsson R, Nordin M. Road traffic noise, air pollution, and risk of dementia – results from the Betula project. Environ Res. Oct 2018;166:334-339. doi:10.1016/j.envres.2018.06.008
- 93. Fania A, Monaco A, Amoroso N, et al. Machine learning and XAI approaches highlight the strong connection between O3 and NO2 pollutants and Alzheimer’s disease. Sci Rep. Mar 5 2024;14(1):5385. doi:10.1038/s41598-024-55439-1
- 94. He F, Tang J, Zhang T, et al. Impact of air pollution exposure on the risk of Alzheimer’s disease in China: A community-based cohort study. Environ Res. Apr 1 2022;205:112318. doi:10.1016/j.envres.2021.112318
- 95. Wood D, Evangelopoulos D, Beevers S, Kitwiroon N, Katsouyanni K. Exposure to Ambient Air Pollution and the Incidence of Dementia in the Elderly of England: The ELSA Cohort. Int J Environ Res Public Health. Nov 29 2022;19(23):16018. doi:10.3390/ijerph192316018
- 96. Trievaan MHJ, Heyworth J, Almeida OP, et al. Ambient air pollution and risk of incident dementia in older men living in a region with relatively low concentrations of pollutants: The Health in Men Study. Environ Res. Dec 2022;215(Pt 2):114349. doi:10.1016/j.envres.2022.114349
- 97. Andersen ZJ, Zhang J, Jorgensen JT, et al. Long-term exposure to air pollution and mortality from dementia, psychiatric disorders, and suicide in a large pooled European cohort: ELAPSE study. Environ Int. Dec 2022;170:107581. doi:10.1016/j.envint.2022.107581
- 98. de Crom TOE, Ginos BNR, Oudin A, Ikram MK, Voortman T, Ikram MA. Air Pollution and the Risk of Dementia: The Rotterdam Study. J Alzheimers Dis. 2023;91(2):603-613. doi:10.3233/JAD-220804
- 99. Blanco MN, Shaffer RM, Li G, et al. Traffic-related air pollution and dementia incidence in the Adult Changes in Thought Study. Environ Int. Jan 2024;183:108418. doi:10.1016/j.envint.2024.108418
- 100. Shim JI, Byun G, Lee JT. Long-term exposure to particulate matter and risk of Alzheimer’s disease: A nationwide population-based Cohort Study. Environ Health. Apr 14 2023;22(1):35. doi:10.1186/s12940-023-00986-9
- 101. Li Z, Christensen GM, Lath JJ, et al. Neighborhood characteristics as confounders and effect modifiers for the association between air pollution exposure and subjective cognitive functioning. Environ Res. Sep 2022;212(Pt A):1132-21. doi:10.1016/j.envres.2022.113221
- 102. McMaughan DJ, Oloruntoba O, Smith ML. Socioeconomic Status and Access to Healthcare: Interrelated Drivers for Healthy Aging. Front Public Health. 2020;8:231. doi:10.3389/fpubh.2020.00231
- 103. Frndak S, Deng Z, Ward-Caviness CK, Gorski-Steiner I, Thorpe RJ, Dickerson AS. Risk of dementia due to Co-exposure to air pollution and neighborhood disadvantage. Environ Res. Jun 15 2024;251(Pt 2):118709. doi:10.1016/j.envres.2024.118709
- 104. Alemany S, Crous-Bou M, Vilor-Tejedor N, et al. Associations between air pollution and plasma biomarkers of Alzheimer’s disease in cognitively unimpaired individuals. Environ Int. Dec 2021;157:106684. doi:10.1016/j.envint.2021.106684
105. Crous-Bou M, Gascon M, Gispert JD, et al. Impact of urban environmental exposures on cognitive performance and brain structure of healthy individuals at risk for Alzheimer’s dementia. Environ Int. May 2020;138:105546. doi:10.1016/j.envint.2020.105546
106. Cho J, Jang H, Park H, et al. Alzheimer’s disease-like cortical atrophy mediates the effect of air pollution on global cognitive function. Environ Int. Jan 2023;171:107703. doi:10.1016/j.envint.2022.107703
107. Falcon C, Gascon M, Molinuevo JL, et al. Brain correlates of urban environmental exposures in cognitively unimpaired individuals at increased risk for Alzheimer’s disease: A study on Barcelona’s population. Alzheimers Dement (Amst). 2021;13(1):e12025. doi:10.1002/dad2.12205
108. Casey E, Li Z, Liang D, et al. Association between Fine Particulate Matter Exposure and Cerebrospinal Fluid Biomarkers of Alzheimer’s Disease in Cognitively Healthy Veterans: A Project on the Epidemiology of Cognitive Health Study Nested Case-Control Based Cohort. Environ Health Perspect. Apr 2024;132(4):47001. doi:10.1289/EHP13503
109. Calderon-Garcidueñas L, Herrera-Soto A, Jury N, et al. Reduced repressive epigenetic marks, increased DNA damage and Alzheimer’s disease hallmarks in the brain of humans and mice exposed to particulate urban air pollution. Environ Res. Apr 2020;183:109226. doi:10.1016/j.envres.2020.109226
110. Calderon-Garcidueñas L, Gonzalez-Maciel A, Reynoso-Robles R, et al. Alzheimer’s disease and alpha-synuclein pathology in the olfactory bulbs of infants, children, teens and adults </= 40 years in Metropolitan Mexico City. APOE4 carriers at higher risk of suicide accelerate their olfactory bulb pathology. Environ Res. Oct 2018;166:348-362. doi:10.1016/j.envres.2018.06.027
111. Shaffer RM, Li G, Adar SD, et al. Fine Particulate Matter and Markers of Alzheimer’s Disease Neuropathology at Autopsy in a Community-Based Cohort. J Alzheimers Dis. 2021;79(4):1716-1731. doi:10.3233/JAD-210053
112. Calderon-Garcidueñas L, Solorio E, Ramirez MC, et al. Inflammatory bowel disease and neurodegenerative diseases, frontotemporal lobar degeneration and amyotrophic lateral sclerosis overlapping neuropathology start in the first two decades of life in pollution exposed urbanites and brain ultrafine particulate matter and industrial nanoparticles, including Fe, Ti, Al, V, Ni, Hg, Co, Cu, Zn, Ag, Pt, Ce, La, Pr and W are key players. Metropolitan Mexico City health crisis is in progress. Front Hum Neurosci. 2023;17:1297467. doi:10.3389/fnhum.2023.1297467
113. Vanbrabant K, Van Dam D, Bongaerts E, et al. Accumulation of Ambient Black Carbon Particles Within Key Memory-Related Brain Regions. JAMA Netw Open. Apr 1 2024;7(4):e245678. doi:10.1001/jamanetworkopen.2024.5678
114. Cullen B, Newby D, Lee D, et al. Cross-sectional and longitudinal analyses of outdoor air pollution exposure and cognitive function in UK Biobank. Sci Rep. Aug 14 2018;8(1):12089. doi:10.1038/s41598-018-30586-8
115. Lee YG, Yoon SJ, Yoon SH, et al. Air pollution is associated with faster cognitive decline in Alzheimer’s disease. Ann Clin Transl Neurol. Jun 2023;10(6):964-973. doi:10.1002/acn3.51779
116. Yang L, Wan W, Yu C, Xuan C, Zheng P, Yan J. Associations between PM(2.5) exposure and Alzheimer’s Disease prevalence among elderly in eastern China. Environ Health. Nov 29 2022;21(1):119. doi:10.1186/s12940-022-00937-w
117. Younan D, Wang X, Millstein J, et al. Air quality improvement and cognitive decline in community-dwelling older women in the United States: A longitudinal cohort study. PLoS Med. Feb 2022;19(2):e1003839. doi:10.1371/journal.pmed.1003839
118. Franz CE, Gustavson DE, Elman JA, et al. Associations Between Ambient Air Pollution and Cognitive Abilities from Midlife to Early Old Age: Modification by APOE Genotype. J Alzheimers Dis. 2023;93(1):193-209. doi:10.3233/JAD-221504
119. Franz CE, Gustavson DE, Elman JA, et al. Associations Between Ambient Air Pollution and Cognitive Abilities from Midlife to Early Old Age: Modification by APOE Genotype. J Alzheimers Dis. 2023;93(1):193-209. doi:10.3233/JAD-221540
120. Ilango SD, Gonzalez K, Gallo L, et al. Long-Term Exposure to Ambient Air Pollution and Cognitive Function Among Hispanic/Latino Adults in San Diego, California. J Alzheimers Dis. 2021;79(4):1489-1496. doi:10.3233/JAD-200766
121. Kulick ER, Elkind MSV, Boehme AK, et al. Long-term exposure to ambient air pollution, APOE-epsilon4 status, and cognitive decline in a cohort of older adults in northern Manhattan. Environ Int. Mar 2020;136:105440. doi:10.1016/j.envint.2019.105440
122. Lo CC, Liu WT, Lu YH, et al. Air pollution associated with cognitive decline by the mediating effects of sleep cycle disruption and changes in brain structure in adults. Environ Sci Pollut Res Int. Jul 2022;29(35):52355-52366. doi:10.1007/s11356-022-19482-7
123. Park SY, Han J, Kim SH, Suk HW, Park JE, Lee DY. Impact of Long-Term Exposure to Air Pollution on Cognitive Decline in Older Adults With Cognitive Impairment. J Alzheimers Dis. Jun 22 2022;88(2):553-563. doi:10.3233/JAD-215120
124. Ard K, Thomas J, Bullock C. Toxic air pollution and cognitive decline: Untangling particulate matter. Health Place. Aug 16 2024;89:103330. doi:10.1016/j.healthplace.2024.103330
125. Wang X, Younan D, Petkus AJ, et al. Ambient Air Pollution and Long-Term Trajectories of Episodic Memory Decline among Older Women in the WHIMS-ECHO Cohort. Environ Health Perspect. Sep 2021;129(9):97009. doi:10.1289/EHP7668
126. Younan D, Wang X, Gruenewald T, et al. Racial/Ethnic Disparities in Alzheimer’s Disease Risk: Role of Exposure to Ambient Fine Particles. J Gerontol A Biol Sci Med Sci. May 5 2022;77(5):977-985. doi:10.1093/gerona/glab231
127. Raichlen DA, Furlong M, Klimentidis YC, et al. Association of Physical Activity with Incidence of Dementia is Attenuated by Air Pollution. Med Sci Sports Exerc. Jul 1 2022;54(7):1131-1138. doi:10.1249/MSS.0000000000002888
128. Tveitanda MH, Heyworth J, Almeida OP, et al. Ambient air pollution and risk of incident dementia in older men living in a region with relatively low concentrations of pollutants: The Health in Men Study. Environ Res. Dec 2022;215(Pt 2):114349. doi:10.1016/j.envres.2022.114349
129. Zhang Y, Fu Y, Guan X, et al. Associations of ambient air pollution exposure and lifestyle factors with incident dementia in the elderly: A prospective study in the UK Biobank. Environ Int. Jul 4 2024;190:108870. doi:10.1016/j.envint.2024.108870
130. Hu HY, Ma YH, Deng YT, et al. Residential greenness and risk of incident dementia: A prospective study of 375,342 participants. Environ Res. Jan 1 2023;216(Pt 3):114703. doi:10.1016/j.envres.2022.114703
131. Astell-Burt T, Navakatikyan MA, Feng X. Urban green space, tree canopy and 11-year risk of dementia in a cohort of 109,688 Australians. Environ Int. Dec 2020;145:106102. doi:10.1016/j.envint.2020.106102
132. Rodriguez-Loureiro L, Gadeyne S, Bauwelinck M, Lefebvre W, Vanpoucke C, Casas L. Long-term exposure to residential greenness and neurodegenerative disease mortality among older adults: a 13-year follow-up cohort study. Environ Health. May 7 2022;21(1):49. doi:10.1186/s12940-022-00863-x
133. Yu Y, Mayeda ER, Paul KC, et al. Traffic-related Noise Exposure and Late-life Dementia and Cognitive Impairment in Mexican-Americans. Epidemiology. Nov 1 2020;31(6):771-778. doi:10.1097/EDE.0000000000001249
134. Fortea J, Pegueroles J, Alcolea D, et al. APOE4 homozygosity represents a distinct genetic form of Alzheimer’s disease. Nat Med. May 2024;30(5):1284-1291. doi:10.1038/s41591-024-02593-7
135. Jansen IE, Savage JE, Watanabe K, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet. Mar 2019;51(3):404-413. doi:10.1038/s41588-018-0311-9
136. Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer’s disease. Neuron. Aug 13 2009;63(3):287-303. doi:10.1016/j.neuron.2009.06.026
137. Klimentidis YC, Raichlen DA, Bea J, et al. Genome-wide association study of habitual physical activity in over 377,000 UK Biobank participants identifies multiple variants including CADM2 and APOE. Int J Obes (Lond). Jun 2018;42(6):1161-1176. doi:10.1038/s41366-018-0120-3
138. Nuzum H, Stickel A, Corona M, Zeller M, Melrose RJ, Wilkins SS. Potential Benefits of Physical Activity in MCI and Dementia. Behav Neurol. 2020;2020:7807856. doi:10.1155/2020/7807856
139. Shi L, Chen SJ, Ma MY, et al. Sleep disturbances increase the risk of dementia: A systematic review and meta-analysis. Sleep Med Rev. Aug 2018;40:4-16. doi:10.1016/j.smrv.2017.06.010
140. Zhang Y, Shen D, Neng GT, et al. Identifying dementia risk in the UK Biobank. Nat Hum Behav. Jul 2023;7(7):1185-1195. doi:10.1038/s41562-023-01585-x
141. Bein KJ, Wallis CD, Silverman JL, Lein PJ, Wexler AS. Emulating Near-Roadway Exposure to Traffic-Related Air Pollution via Real-Time Emissions from a Major Freeway Tunnel System. Environ Sci Technol. Mar 2 2022;doi:10.1021/acs.est.1c07047
142. Shang Y, Sun Q. Particulate air pollution: major research methods and applications in animal models. Environ Dis. Jul–Sep 2018;3(3):57-62. doi:10.4103/ed.ed_16_18
143. Bendtsen KM, Gren L, Malmborg VB, et al. Particle characterization and toxicity in C57BL/6 mice following instillation of five different diesel exhaust particles designed to differ in physicochemical properties. Part Fibre Toxicol. Aug 8 2020;17(1):38. doi:10.1186/s12989-020-00369-9
144. Moon Y, Park MK, Leikauf GD, Park CS, Jang AS. Diesel exhaust particulates and asthma. Clin Exp Allergy. Jan–Feb 2014;33(1):21-8. doi:10.1177/10915813518355
145. Daniel S, Phillippi D, Schneider LJ, Nguyen KN, Mirpuri J, Lund AK. Exposure to diesel exhaust particles results in altered lung microbial profiles, associated with increased reactive oxygen species/reactive nitrogen species and inflammation, in C57BL/6 wildtype mice on a high-fat diet. Part Fibre Toxicol. Jan 8 2021;18(1):3. doi:10.1186/s12989-020-00393-9
146. Zavala J, Freedman AN, Szilagyi JT, et al. New Approach Methods to Evaluate Health Risks of Air Pollutants: Critical Design Considerations for In Vitro Exposure Testing. Int J Environ Res Public Health. Mar 23 2020;17(6):doi:10.3390/ijerph17062124
147. Lakatos HF, Burgess HA, Thatcher TH, et al. Oropharyngeal aspiration of a silica suspension produces a superior model of silicosis in the mouse. Methods Mol Biol. Jun 2006;32(5):189-95. doi:10.1080/9021406008071465
148. Silva RM, Doudrick K, Franzi LM, et al. Instillation versus inhalation of multiwalled carbon nanotubes: exposure-related health effects, clearance, and the role of particle characteristics. ACS Nano. Sep 23 2014;8(9):8911-31. doi:10.1021/nn503887r
149. Saveleva L, Vartiainen P, Gorova V, et al. Subacute inhalation of ultrafine particulate matter triggers inflammation without altering amyloid beta load in 5xFAD mice. Neurotoxicology. Mar 2022;89:55-66. doi:10.1016/j.neuro.2022.01.001
150. Greve HJ, Dunbar AL, Lombo CG, et al. The bidirectional lung–brain axis of amyloid-beta pathology: ozone dysregulates the peri-plaque microenvironment. Brain. Mar 1 2023;146(3):991-1005. doi:10.1093/brain/awac113
151. Ahmed C, Greve HJ, Garza-Lombo C, et al. Peripheral HMGB1 is linked to O(3) pathogenetic disease-associated alterations of amyloid plaques. Alzheimers Dement. Nov 24 2023;20(5):3551-3566.
- Park SJ, Lee J, Lee S, et al. Exposure of ultrafine particulate matter causes glutathione redox imbalance in the hippocampus: A neurometabolic susceptibility to Alzheimer’s pathology. Sci Total Environ. May 20 2020;718:137267. doi:10.1016/j.scitotenv.2020.137267
- Rivas-Arancibia S, Rodriguez-Martinez E, Badillo-Ramirez I, Lopez-Gonzalez U, Saniger JM. Structural Changes of Amyloid Beta in Hippocampus of Rats Exposed to Ozone: A Raman Spectroscopy Study. Front Mol Neurosci. 2017;10:137. doi:10.3389/fnmol.2017.00137
- Rivas-Arancibia S, Guevara-Guzman R, Lopez-Vidal Y, et al. Oxidative stress caused by ozone exposure induces loss of brain repair in the hippocampus of adult rats. Toxicol Sci. Jan 2010;113(1):187–97. doi:10.1093/toxsci/kfp252
- Jew K, Herr D, Wong C, et al. Selective memory and behavioral alterations after ambient ultrafine particulate matter exposure in aged 3xTgAD Alzheimer’s disease mice. Part Fibre Toxicol. Nov 26 2019;16(1):45. doi:10.1186/s12989-019-0323-3
- Herr D, Jew K, Wong C, et al. Effects of concentrated ambient ultrafine particulate matter on hallmarks of Alzheimer’s disease in the 3xTgAD mouse model. Neurotoxicology. May 2021;84:172–183. doi:10.1016/j.neuro.2021.03.010
- Kilian JG, Mejias-Ortega M, Hsu HW, et al. Exposure to quasi-ultrafine particulate matter accelerates memory impairment and Alzheimer’s disease-like neuropathology in the AppNL-G-F knock-in mouse model. Toxicol Sci. May 31 2023;193(2):175–191. doi:10.1093/toxsci/kfad036
- Sahu B, Mackos AR, Floden AM, Wold LE, Combs CK. Particulate Matter Exposure Exacerbates Amyloid-beta Plaque Deposition and Gliosis in APP/PS1 Mice. J Alzheimers Dis. 2021;80(2):761–774. doi:10.3233/JAD-200919
- Jung HA, C.; Zhao, Y.; Paulson, S.; Anastasio, C.; Wexler, A. Impact of the Versatile Aerosol Concentration Enrichment System (VACES) on Gas Phase Species. Aerosol Science and Technology. 2010;44(12):1113–1121. doi:https://doi.org/10.1080/02786826.2010.512028
- Patten KT, Valenzuela AE, Wallis C, et al. The Effects of Chronic Exposure to Ambient Traffic-Related Air Pollution on Alzheimer’s Disease Phenotypes in Wildtype and Genetically Predisposed Male and Female Rats. Environ Health Perspect. May 2021;129(5):57005. doi:10.1289/EHP8905
- Lee SH, Chen YH, Chien CC, et al. Three-month inhalation exposure to low-level PM2.5 induced brain toxicity in an Alzheimer’s disease mouse model. PLoS One. 2021;16(8):e0254587. doi:10.1371/journal.pone.0254587
- Patten KT, Valenzuela AE, Wallis C, et al. Hippocampal but Not Serum Cytokine Levels Are Altered by Traffic-Related Air Pollution in TgF344-AD and Wildtype Fischer 344 Rats in a Sex- and Age-Dependent Manner. Front Cell Neurosci. 2022;16:861733. doi:10.3389/fncel.2022.861733
- Fu P, Zhao Y, Cheng Q, Cai Z, Li N, Yang Z. SEMA4B upregulation and glial activation enhance Sema4b expression in the brains of Alzheimer’s disease transgenic mice after real-world PM2.5 exposure. J Environ Sci (China). Dec 2022;122:25–40. doi:10.1016/j.jes.2021.02.007
- Ehsanifar M, Yaavari Z, Rafati M. Exposure to urban air pollution particulate matter: neurobehavioral alteration and hippocampal inflammation. Environ Sci Pollut Res Int. Jul 2022;29(33):50856–50866. doi:10.1007/s11356-022-19367-9
- Zhang W, Xiao D, Mao Q, Xia H. Role of neuroinflammation in neurodegeneration development. Signal Transduct Target Ther. Jul 12 2023;8(1):267. doi:10.1038/s41392-023-01846-5
- Hopperton KE, Mohammad D, Trepanier MO, Giuliano V, Bazinet RP. Markers of microglia in post-mortem brain samples from patients with Alzheimer’s disease: a systematic review. Mol Psychiatry. Feb 2018;23(2):177–198. doi:10.1038/mp.2017.246
- Weverling JW, Geut H, Bol JH, et al. Neuroinflammation is associated with Alzheimer’s disease co-pathology in dementia with Lewy bodies. Acta Neuropathol Commun. May 7 2024;12(1):73. doi:10.1186/s40478-024-01786-z
- Liu WS, Zhang YR, Ge YJ, Wang HF, Cheng W, Yu JT. Inflammation and Brain Structure in Alzheimer’s Disease and Other Neurodegenerative Disorders: a Mendelian Randomization Study. Mol Neurobiol. Mar 2024;61(3):1593–1604. doi:10.1007/s12035-023-03648-6
- Rangaraju S, Dammer EB, Raza SA, et al. Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol Neurodegener. May 21 2018;13(1):24. doi:10.1186/s13024-018-0254-8
- Saito T, Saido TC. Neuroinflammation in mouse models of Alzheimer’s disease. Clin Exp Neuroimmunol. Nov 2018;9(4):211-218. doi:10.1111/cen3.12475
- Mucke L, Selkoe DJ. Neurotoxicity of amyloid beta-protein: synaptic and network dysfunction. Cold Spring Harb Perspect Biol. Jul 2012;4(7):a006338. doi:10.1101/cshperspect.a006338
- Chen Y, Yu Y, Tau and neuroinflammation in Alzheimer’s disease: interplay mechanisms and clinical translation. J Neuroinflammation. Jul 14 2023;20(1):165. doi:10.1186/s12974-023-02853-3
- Rauti R, Cellot G, D’Andrea P, et al. BDNF impact on synaptic dynamics: extra or intracellular long-term release differently regulates cultured hippocampal synapses. Mol Brain. Mar 17 2020;13(1):43. doi:10.1186/s13041-020-00582-9
- Stevens B, Allen NJ, Vazquez LE, et al. The classical complement cascade mediates CNS synapse elimination. Cell. Dec 14 2007;131(6):1164–78. doi:10.1016/j.cell.2007.10.036
- Jantti H, Sitnikova V, Ishchenko Y, et al. Microglial amyloid beta clearance is driven by PIEZO1 channels. J Neuroinflammation. Jun 15 2022;19(1):147. doi:10.1186/s12974-022-02486-y
- Zhan Z, Jalabi W, Shpargel KB, et al. Lipopolysaccharide-induced microglial activation and neuroprotection against experimental brain injury is independent of hematopoiesis TLR4. J Neurosci. Aug 22 2012;32(34):11706–15. doi:10.1523/JNEUROSCI.0730-12.2012
- Fan J, Dong X, Tang Y, et al. Preferential pruning of inhibitory synapses by microglia contributes to alteration of the balance between excitatory and inhibitory synapses in the hippocampus in temporal lobe epilepsy. CNS Neurosci Ther. Oct 2023;29(10):2884–2900. doi:10.1111/cns.14224
- Taddei RN, Perbet R, Mate de Gerando A, et al. Tau Oligomer-Containing Synapse Elimination by Microglia and Astrocytes in Alzheimer Disease. JAMA Neurol. Nov 1 2023;80(11):1209-1221. doi:10.1001/jamaneurol.2023.3530
- Chung HY, Wickel J, Hahn N, et al. Microglia mediate neurocognitive deficits by eliminating C1q-tagged synapses in sepsis-associated encephalopathy. Sci Adv. May 26 2023;9(21):eabq7806. doi:10.1126/sciadv.abq7806
- Jha SJ, Fang XH, Li YJ, Deng Y, Wei WS, Zhang Y. X. Z-SF1/simHIF-3α signaling following hypoxia/ischemia. Mol Biol Rep. Aug 2022;49(8):7697-7707. doi:10.1007/s11033-022-07587-8
- Liang P, Zhang X, Zhang Y, et al. Neurotoxic A1 astrocytes promote neuronal ferroptosis via CXCL10/CXCR3 axis in epilepsy. Free Radic Biol Med. Feb 1 2023;195:329-342. doi:10.1016/j.freeradbiomed.2023.01.002
- Hong S, Beja-Glasser VF, Nfonoyim BM, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. May 6 2016;352(6286):712-716. doi:10.1126/science.aad8373
- Imai Y, Ibata I, Ito D, Ohsawa K, Kohsaka S. A novel gene iba1 in the major histocompatibility complex class III region encoding an EF hand protein expressed in a monocytic lineage. Biochem Biophys Res Commun. Jul 25 1996;224(3):855-62. doi:10.1006/bbrc.1996.1195
- Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S. Microglia-specific localization of a novel calcium binding protein, Iba1. Brain Res Mol Middeldorp J, Hol EM. GFAP in health and disease. Prog Neurobiol. Mar 2011;93(3):421–43. doi:10.1016/j.pneurobio.2011.01.005
- Eng LF. Glial fibrillary acidic protein (GFAP): the major protein of glial intermediate filaments in differentiated astrocytes. J Neuroimmunol. Jun 1985;8(4-6):203–14. doi:10.1016/s0165-5728(85)80063-1
- Ji X, Liu R, Guo J, et al. Olfactory bulb microglia activation mediated neuronal death in real-ambient particulate matter exposure mice with depression-like behaviors. Sci Total Environ. May 15 2022;821:153456. doi:10.1016/j.scitotenv.2022.153456
- Jang S, Kim EW, Zhang Y, et al. Particulate matter increases beta-amyloid and activated glial cells in hippocampal tissues of transgenic Alzheimer’s mouse: Involvement of PPAR-γ. Biochem Biophys Res Commun. Jun 2 2018;500(2):333–338. doi:10.1016/j.bbrc.2018.04.068
- Xu MX, Zhu YF, Chang HF, Liang Y. Nanoceria restrains PM2.5-induced metabolic disorder and hypothalamus inflammation by inhibition of astrocytes activation related NF-kappaB pathway in Nrf2 deficient mice. Free Radic Biol Med. Oct 2016;99:259–272. doi:10.1016/j.freeradbiomed.2016.08.021
- Andrade-Oliva MD, Atzati-Aguilar OG, Garcia-Sierra F, De Vizcaya-Ruiz A, Arias-Montano JA. Effect of in vivo exposure to ambient fine particles (PM2.5)) on the density of dopamine D(2)-like receptors and dopamine-induced ([35S]GTPgammaS binding in rat prefrontal cortex and striatum membranes. Environ Toxicol Pharmacol. Jun 2018;60:58–65. doi:10.1016/j.etap.2018.04.001
- Zhong L, Sheng X, Wang W, et al. TREM2 receptor protects against complement-mediated synaptic loss by binding to complement C1q during neurodegeneration. Immunity. Aug 8 2023;56(8):1794–1808 e8. doi:10.1016/j.immuni.2023.01.006
- Saha P, Sarkar S, Paidi RK, Biswas SC. TIMP-1: A key cytokine released from activated astrocytes protects neurons and ameliorates cognitive behaviours in a rodent model of Alzheimer’s disease. Brain Behav Immun. Jul 2020;87:804–819. doi:10.1016/j.bbi.2020.03.014
- Chen Z, Jalabi W, Hu W, et al. Microglial displacement of inhibitory synapses provides neuroprotection in the adult brain. Nat Commun. Jul 22 2014;5:4486. doi:10.1038/ncomms5486
- Li B, Chang X, Liang X, et al. The role of reactive astrocytes in neurotoxicity induced by ultrafine particulate matter. Sci Total Environ. Apr 1 2023;867:161416. doi:10.1016/j.scitotenv.2023.161416
- Kang YJ, Tan HY, Lee CY, Cho H. An Air Particulate Pollutant Induces Neuroinflammation and Neurodegeneration in Human Brain Models. Adv Sci (Weinh). Nov 2021;8(21):e2101251. doi:10.1002/advs.202101251
- Bouter YF. Amyloid-beta 1-42 induced hippocampal neurodegeneration. Alzheimers Dement. Sep 2023;19(9):4237–4247. doi:10.1002/alz.13165
- Bai R, Guo J, Ye XY, Xie Y, Xie T. Oxidative stress: The core pathogenesis and mechanism of Alzheimer’s disease. Ageing Res Rev. May 2022;77:101619. doi:10.1016/j.arr.2022.101619
- Vilhardt F, Haslund-Vinding J, Jaquet V, McBean G. Microglia antioxidant systems and redox signalling. Br J Pharmacol. Jun 2017;174(12):1719–1732. doi:10.1111/bph.13426
- Suwannasual U, Lucero J, McDonald JD, Lund AK. Exposure to traffic-generated air pollutants mediates alterations in brain microvascular integrity and cognitive function in mice on a high-fat diet. Environ Res. Jan 2018;160:449–461. doi:10.1016/j.envres.2017.10.029
- Ehsanifar M, Montazeri Z, Taheri MA, Rafati M, Behjati M, Karimian H. Hippocampal inflammation and oxidative stress following exposure to diesel exhaust particles in male and female mice. Neurochem Int. May 2022;154:105284. doi:10.1016/j.neuint.2021.104989
Ehsanifar M, Montazeri Z, Taheri MA, Rafati M, Behjati M, Karimian H. Hippocampal inflammation and oxidative stress following exposure to diesel exhaust particles in male and female mice. Neurochem Int. May 2022;154:105284. doi:10.1016/j.neuint.2021.104989 Cacciottolo M, Morgan TE, Saffari AA, et al. Traffic-related air pollutants (TRAP-PM) promote neuronal amyloidogenesis through oxidative damage to lipid rafts. Free Radic Biol Med. Feb 1 2020;147:242–251. doi:10.1016/j.freeradbiomed.2019.12.023
Lee SH, Lin CY, Chen TF, et al. Distinct brain lipid signatures in response to low-level PM(2.5) exposure in a 3xTg-Alzheimer’s disease mouse inhalation model. Sci Total Environ. Sep 10 2022;838(Pt 4):156456. doi:10.1016/j.scitotenv.2022.156456
Bai KJ, Chuang KJ, Wu SM, et al. Effects of diesel exhaust particles on the expression of tau and autophagy proteins in human neuroblastoma cells. Environ Toxicol Pharmacol. Sep 2018;62:54–59. doi:10.1016/j.etap.2018.06.007
Milani C, Corsetto PA, Farina F, et al. Early evidence of stress in immortalized neurons exposed to diesel particles: the role of lipid peroxidation in cognitive stress and inflammation. Toxicology. Nov 2010;276(1):22–28. doi:10.1016/j.tox.2010.07.015
Wang GY, Shi SZ, Ge NN, et al. PM2.5 exposure aggravates oligomeric amyloid beta-induced neuronal injury and promotes NLRP3 inflammasome activation in an in vitro model of Alzheimer’s disease. J Neuroinflammation. May 2 2018;15(1):132. doi:10.1186/s12974-018-1178-5
Aquino GV, Dabi A, Odom GJ, Zhang F, Bruce ED. Evaluating the endothelial-microglial interaction and comprehensive inflammatory marker profiles under acute exposure to ultrafine diesel exhaust particles in vitro. Toxicology. Apr 30 2021;454:152748. doi:10.1016/j.tox.2021.152748
Shi Q, Chowdhury S, Ma R, et al. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci Transl Med. May 31 2017;9(392):doi:10.1126/scitranslmed.aaf6295
Zhang J, Malik A, Choi HB, Ko RW, Dissing-Olesen L, MacVicar BA. Microglial CR3 activation triggers long-term synaptic depression in the hippocampus via NADPH oxidase. Neuron. Apr 2 2014;82(1):195–207. doi:10.1016/j.neuron.2014.01.043
Hou L, Wang K, Zhang C, et al. Complement receptor 3 mediates NADPH oxidase activation and dopaminergic neurodegeneration through a Src-Erk–dependent pathway. Redox Biol. Apr 2018;14:250–260. doi:10.1016/j.redox.2017.09.017
Lian H, Litvinchuk A, Chiang AC, Aithmitti N, Jankowsky JL, Zheng H. Astrocyte-Microglia Cross Talk through Complement Activation Modulates Amyloid Pathology in Mouse Models of Alzheimer’s Disease. J Neurosci. Jan 13 2016;36(2):577–89. doi:10.1523/JNEUROSCI.2117-15.2016
Kumar A, Barrett JP, Alvarez-Croda DM, Stoica BA, Faden AI, Loane DJ. NOX2 drives M1-like microglial/macrophage activation and neurodegeneration following experimental traumatic brain injury. Brain Behav Immun. Nov 2016;58:291–309. doi:10.1016/j.bbi.2016.07.158
Zhang H, Haghani A, Mousavi AH, et al. Cell-based assays that predict in vivo neurotoxicity of urban ambient nano-sized particulate matter. Redox Biol Med. Dec 2019;145:33–41. doi:10.1016/j.freeradbiomed.2019.09.009
Velazquez-Perez YE, Rodriguez-Martinez Y, Valdes-Fuentes M, Gelista-Herrera N, Gomez-Crisostomo N, Rivas-Arancibia S. Oxidative Stress Caused by Ozone Exposure Induces Changes in P2X7 Receptors, Neuroinflammation, and Neurodegeneration in the Rat Hippocampus. Oxid Med Cell Longev. 2021;2021:3790477. doi:10.1155/2021/3790477
Solleiro-Villavicencio H, Hernandez-Orozco E, Rivas-Arancibia S. Effect of exposure to low doses of ozone on interleukin 17A expression during progressive neurodegeneration in the rat hippocampus. Neurologia (Engl Ed). Nov–Dec 2021;36(9):673–680. doi:10.1016/j.nrleng.2018.08.003
Singh J, Habeam ML, Panicker N. Inflammosome assembly in neurodegenerative diseases. Trends Neurosci. Oct 2023;46(10):814–831. doi:10.1016/j.tins.2023.07.009
Bourganis V, Kammon A, Olexopoulos A, Kiparissides C. Recent advances in nose-to-brain delivery of pharmaceuticals. Eur J Pharm Biopharm. Apr 2018;128:337–362. doi:10.1016/j.ejpb.2018.05.009
Ajmani GS, Suh HH, Pinto JM. Effects of Ambient Air Pollution Exposure on Olfaction: A Review. Environ Health Perspect. Nov 2016;124(11):1683–1693. doi:10.1289/EHP136
Gartzianidia O, Egusquizaquirre SP, Bianco J, et al. Nanoparticle transport across in vitro olfactory cell monolayers. Int J Pharm. Feb 29 2016;499(1–2):81–89. doi:10.1016/j.ijpharm.2015.12.046
Elder A, Gelein R, Silva V, et al. Translocation of inhaled ultrafine manganese oxide particles to the central nervous system. Environ Health Perspect. Aug 2006;114(8):1172–8. doi:10.1289/ehp.9030
Liang C, Jiang Y, Zhang T, et al. Atmospheric particulate matter impairs cognition by modulating synaptic function via the nose-to-brain route. Sci Total Environ. Jan 20 2023;857(Pt 3):159600. doi:10.1016/j.scitotenv.2022.159600
Kilian J, Kitazawa M. The emerging risk of exposure to air pollution on cognitive decline and Alzheimer’s disease – Evidence from epidemiological and animal studies. Biomed J. Jun 2018;41(3):141-162. doi:10.1016/j.bj.2018.06.001
Wang W, Lin Y, Yang H, et al. Internal Exposure and Distribution of Airborne Fine Particles in the Human Body: Methodology, Current Understandings, and Research Needs. Environ Sci Technol. Jun 7 2022;56(11):6857–6869. doi:10.1021/acs.est.1c07051
You R, Ho YS, Chang RC. The pathogenic effects of particulate matter on neurodegeneration: a review. J Biomed Sci. Feb 22 2022;29(1):15. doi:10.1186/s12929-022-00799-x
Calderon-Garcidueñas L, Perez-Calatayud AA, Gonzalez-Maciel A, et al. Environmental Nanoparticles Reach Human Fetal Brains. Biomedicines. Feb 9 2022;10(2) doi:10.3390/biomedicines10020410
Li Y, Liu Y, Hu C, et al. Study of the neurotoxicity of indoor airborne nanoparticles based on a 3D human blood-brain barrier chip. Environ Int. Oct 2020;143:105598. doi:10.1016/j.envint.2020.105598
Yamazaki Y, Shinohara M, Shinohara M, et al. Selective loss of cortical endothelial tight junction proteins during Alzheimer’s disease progression. Brain. Apr 1 2019;142(4):1077–1092. doi:10.1093/brain/awz011
Kurz C, Walker L, Rauchmann BS, Perneczky R. Dysfunction of the blood-brain barrier in Alzheimer’s disease: Evidence from human studies. Neuropathol Appl Neurobiol. Apr 2022;48(3):e12782. doi:10.1111/nan.12782
Cai Z, Qiao PF, Wan CQ, Cai M, Zhou NK, Li Q. Role of Blood-Brain Barrier in Alzheimer’s Disease. J Alzheimers Dis. 2018;63(4):1223–1234. doi:10.3233/JAD-180098
Bernardino PN, Hobson BA, Huddleston SL, et al. Time- and region-dependent blood-brain barrier impairment in a rat model of organophosphate-induced status epilepticus. Neurobiol Dis. Oct 15 2023;187:106316. doi:10.1016/j.nbd.2023.106316
Park JH, Choi JY, Lee HK, Jo C, Koh YH. Notch1-mediated inflammation is associated with blood–brain barrier dysfunction in human microvascular endothelial cells upon particulate matter exposure. Arch Toxicol. Feb 2021;95(2):529–540. doi:10.1007/s00204-020-02942-9
Adivi A, Lucero J, Simpson N, McDonald JD, Lund AK. Exposure to traffic-generated air pollution promotes alterations in the integrity of the brain microvasculature and inflammation in female ApoE(-/-) mice. Toxicol Lett. Mar 15 2021;339:39–50. doi:10.1016/j.toxlet.2020.12.016
Roque PJ, Dao K, Costa LG. Microglia mediate diesel exhaust particle-induced cerebellar neuronal toxicity through neuroinflammatory mechanisms. Neurotoxicology. Sep 2016;56:204–214. doi:10.1016/j.neuro.2016.08.006
Han B, Li X, Ai RS, et al. Atmospheric particulate matter aggravates CNS demyelination through microglial TLR4/NF-κB signaling and microglial activation. Elife. Feb 24 2022;11 doi:10.7554/eLife.72247
Pradhan SH, Gibb M, Kramer AT, Sayes CM. Peripheral (ungroomed) exposure to diesel-engine exhaust inhalation induces oxidative stress and