

ALS Treatment Using Bone Marrow-Derived Extracellular Vesicles
Case Report: Amyotrophic Lateral Sclerosis Treatment with Extracellular Vesicles derived from Mesenchymal Stromal Cells
Paul A. Dreschnack, MD¹; Ina Dreschnack, MS²; Robert L. Bard, MD³
- Paul A. Dreschnack, MD, P.C.
- Harvard University
- Bard Cancer Center
OPEN ACCESS
PUBLISHED: 30 September 2025
CITATION: Dreschnack, PA., Dreschnack, I., et al., 2025. Case Report: Amyotrophic Lateral Sclerosis Treatment with Extracellular Vesicles derived from Mesenchymal Stromal Cells. Medical Research Archives, [online] 13(9). https://doi.org/10.18103/mra.v13i9.6930
COPYRIGHT: This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
DOI https://doi.org/10.18103/mra.v13i9.6930
ISSN 2375-1924
ABSTRACT
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disorder marked by motor neuron loss, resulting in muscle weakness, respiratory failure, and death. Current treatment options offer limited survival benefit, highlighting the urgent need for novel therapeutic strategies. Extracellular vesicles (EVs), particularly those derived from human bone marrow, have shown promise in preclinical models due to their anti-inflammatory and neuroprotective properties.
We report the case of an 81-year-old male with a two-year history of ALS who received a novel investigational therapy consisting of intravenous infusions of human bone marrow-derived EVs. At baseline, the patient exhibited bulbar and respiratory involvement, as well as progressive weakness in the upper and lower extremities, and significant impairment in activities of daily living. Pulmonary function testing revealed a forced vital capacity (FVC) of 52% predicted, and the patient relied on non-invasive ventilation for up to 9 hours nightly.
The treatment was well-tolerated with no adverse events reported. Clinical monitoring showed stabilization of respiratory function, maintained ambulatory ability for short distances, and no further significant decline in bulbar or limb strength over the treatment period. Subjective improvements were noted in energy levels, mood, and overall quality of life, though these outcomes require further investigation in controlled studies.
Keywords
Amyotrophic lateral sclerosis, extracellular vesicles, mesenchymal stromal cells, neurodegenerative diseases, treatment
Introduction
Amyotrophic lateral sclerosis (ALS) is a relentlessly progressive neurodegenerative disorder characterized by the degeneration of upper and lower motor neurons, leading to progressive muscle weakness, respiratory compromise, and eventual death, typically within 3 to 5 years of symptom onset. It affects an estimated 6.6 persons per 100,000 individuals in the US. Despite ongoing research, current therapeutic options for ALS remain limited and offer only modest survival benefits. Consequently, there is a critical unmet need for novel interventions that can alter the course of the disease or improve patients’ quality of life.
In this case report, we describe the clinical course of an 81-year-old male with a two-year history of ALS who underwent a novel treatment protocol involving intravenous administration of human bone marrow-derived EVs. This report outlines the patient’s baseline neurological and functional status, the treatment regimen, and the observed outcomes over five months. In this case report study, we hypothesized that IV administration of bone marrow–derived extracellular vesicles (EVs) could achieve some degree of improvement in the patient’s basic daily life needs, even if it did not reverse the underlying disease pathomechanism of amyotrophic lateral sclerosis (ALS). We anticipated that EV therapy might provide functional stabilization or modest enhancement in quality-of-life measures, such as mobility, speech, and self-care.
However, given the patient’s advanced age and the accelerated onset of disease progression, we considered these factors as potential limitations that could inhibit major improvements at the level of disease pathophysiology. Accordingly, our expectations focused on achieving functional gains and maintaining independence, rather than reversing the neurodegenerative process itself.
In addition, the study aimed to assess the tolerability and safety of repeated EV-based therapy infusions administered over five months in an ALS patient, and to contribute to the growing body of literature by examining the potential limitations of EV therapy in the context of advanced age. They accelerated disease progression, acknowledging their likely impact on the scope of therapeutic benefit. Recent advances in regenerative medicine have identified extracellular vesicles (EVs), including exosomes and microvesicles, as promising therapeutic agents in neurodegenerative diseases. EVs are nanoscale, membrane-bound particles released by cells that facilitate intercellular communication and modulate immune responses, inflammation, and tissue repair. Preclinical studies have demonstrated that EVs derived from mesenchymal stromal cells (MSCs), particularly those of human bone marrow origin, may have neuroprotective and anti-inflammatory effects, potentially mitigating disease progression in ALS.
Method: Case report
Our patient was an 81-year-old male, a retired philosophy professor, with a two-year history of amyotrophic lateral sclerosis (ALS) before initiation of this treatment protocol. His past medical history was notable for benign prostatic hyperplasia, coronary artery disease with prior stent placement, gastroesophageal reflux disease, hypothyroidism, hyperlipidemia, and testicular hypogonadism. At the time of evaluation, his medication regimen given by his primary physician included aspirin, atorvastatin (Lipitor), azelastine (Astelin), clonazepam (Klonopin), vitamin B12, famotidine (Pepcid), fexofenadine (Allegra), guaifenesin (Mucinex), levothyroxine (Synthroid), omega-3 fatty acids, riluzole (Rilutek), and tamsulosin (Flomax).
A neurological assessment conducted three months before treatment reported mild facial and lingual weakness with an intact gag reflex. The patient exhibited moderate dysphagia, characterized by occasional coughing and choking on saliva and pills, though he tolerated solids and liquids without significant difficulty. Progressive respiratory involvement was noted, including orthopnea and exertional dyspnea, which occurred during routine activities such as speaking, chewing, and eating. Pulmonary function testing revealed a forced vital capacity (FVC) of 52% predicted, indicating significant weakness of the respiratory muscles. The patient was unable to lie flat and demonstrated hypophonia and ineffective cough. He was utilizing non-invasive ventilation (NIV) for approximately 2 hours during the day and 8–9 hours nightly.
Motor examination of the upper extremities revealed progressive weakness (more pronounced on the left and in distal muscles), intrinsic hand atrophy, fasciculations, and slowed rapid alternating movements bilaterally. Bilateral first dorsal interosseus muscle atrophy resulted in a total lack of thumb opposition. Lower extremity findings included marked atrophy, fasciculations, and severely impaired rapid movements. The patient initially developed a left foot drop accompanied by progressive gait instability and falls, which preceded other neurological symptoms. Within a year of onset, he required a walker, and by six weeks before diagnosis, he was wheelchair-bound. Despite this, he remained ambulatory over short distances (approximately 40 feet) to and from the bathroom using a walker, which he accomplished approximately four times daily. Transfers to and from the wheelchair required the use of his upper limbs for support.
The patient was engaged in a regular home-based physical therapy and exercise program. Nutritional assessment indicated adequate oral intake, although the patient reported a long-term weight decline from 200 lbs. to 152 lbs. over the previous decade, attributed to anorexia and progressive muscle wasting. Psychiatric evaluation revealed the presence of anxiety without evidence of depression, delusions, or hallucinations. Overall, the patient experienced increasing difficulty with basic activities of daily living due to the progressive nature of his neuromuscular impairments.
Ultrasonic studies performed by Dr. Bard showed very limited diaphragmatic excursion (1cm) at rest. Orbital studies indicated normal orbital examination, except for reduced diameter optic nerves. Papilledema was not present. A repeat diaphragmatic study post-treatment did not show any improvement in the excursion of the diaphragm.
In this clinical case report, we administered a series of treatments utilizing extracellular vehicles (EVs) of human origin, derived from freshly harvested human bone marrow. The treatment protocol consisted of a total of 15 intravenous (IV) administrations administered over five months. Each treatment involved the IV infusion of 15 cc of EVs. The treatments were scheduled for three alternating days during one week of each month. The Esch infusion consisted of 15cc of Direct Biologics ExoFlo. We used the ALS Functional Rating Scale Revised with the Neurology Tool Kit to evaluate the severity of ALS present before and after treatment. Crose in his article suggested that EVs could successfully delay the progression of the disease process.
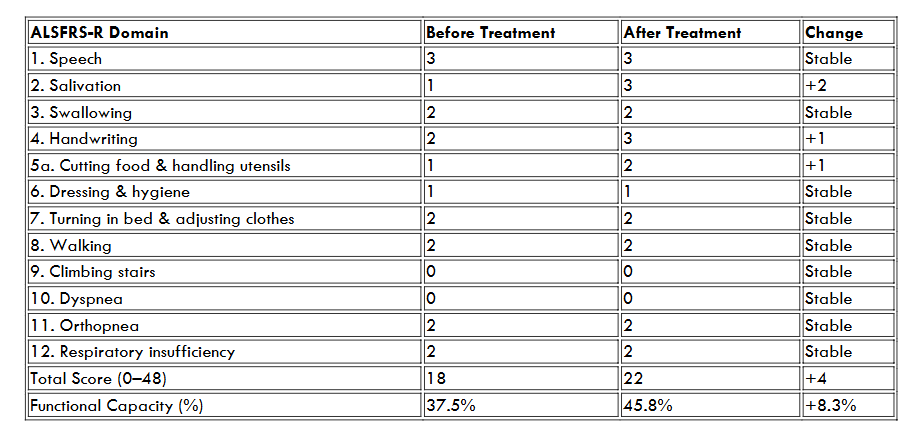
The ALS Functional Rating Scale–Revised (ALSFRS-R) was administered to a patient with amyotrophic lateral sclerosis (ALS) prior to and following administration of extracellular vesicle (EV) therapy. The ALSFRS-R is a validated 12-item instrument widely used to assess functional impairment in ALS, with scores ranging from 0 (maximal disability) to 48 (no impairment). Before treatment, the patient’s initial ALSFRS-R score was 18/48 (37.5%), reflecting moderate-to-severe functional impairment. Major deficits were observed in salivation (1/4), cutting food and handling utensils (1/4), dressing and hygiene (1/4), climbing stairs (0/4), and dyspnea (0/4). These findings indicate pronounced bulbar dysfunction, loss of independence in activities of daily living, and significant respiratory compromise. Following EV administration, the patient’s ALSFRS-R score improved to 22/48 (45.8%), corresponding to a 4-point (8.3%) functional gain. Improvements were most notable in salivation (increased from 1 to 3 points, suggesting reduced drooling and better oral motor control), handwriting (increased from 2 to 3 points, indicating improved fine motor dexterity), and cutting food/handling utensils: increased from 1 to 2 points, reflecting partial recovery of self-feeding ability. Other domains, including speech, swallowing, ambulation, respiratory status, and dressing/hygiene, remained stable with no evidence of decline over the treatment period.


One of the most notable changes after therapy with extracellular vesicles was the restoration of thumb opposition. Before initiating treatment, he totally lacked thumb opposition. In transferring from his bed, he could use his index through his little fingers to grasp an overhead transfer bar. With restoration, he was able to use his hands more effectively for transfer.
Another observation we noted was an increase in his ability to ambulate. The distance increased by about 100 paces without assistance. Thus, he could ambulate in his apartment without being completely dependent on his walker. This increase in his capability facilitated his restoration of bathroom privileges, reducing his need for bedside accommodation.
| ALSFRS-R Domain | Before Treatment | After Treatment | Change |
|---|---|---|---|
| 1. Speech | 3 | 3 | Stable |
| 2. Salivation | 1 | 3 | +2 |
| 3. Swallowing | 2 | 2 | Stable |
| 4. Handwriting | 2 | 3 | +1 |
| 5a. Cutting food & handling utensils | 1 | 2 | +1 |
| 6. Dressing & hygiene | 1 | 1 | Stable |
| 7. Turning in bed & adjusting clothes | 2 | 2 | Stable |
| 8. Walking | 2 | 2 | Stable |
| 9. Climbing stairs | 0 | 0 | Stable |
| 10. Dyspnea | 0 | 0 | Stable |
| 11. Orthopnea | 2 | 2 | Stable |
| 12. Respiratory insufficiency | 2 | 2 | Stable |
| Total Score (0–48) | 18 | 22 | +4 |
| Functional Capacity (%) | 37.5% | 45.8% | +8.3% |
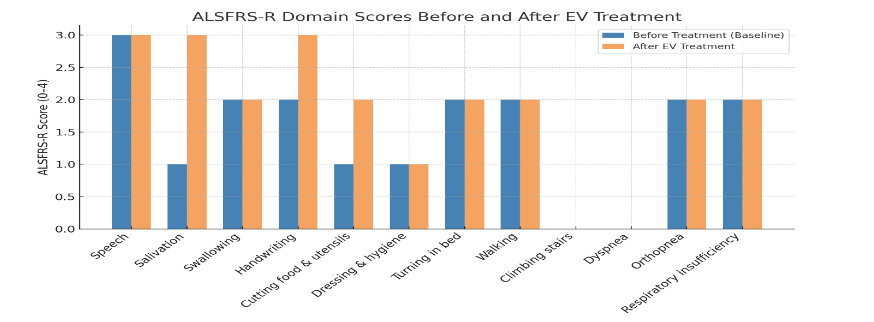
Notably, the restoration of thumb opposition is suggestive that some motor recovery is possible and sustainable. These findings suggest that EV therapy may provide clinically meaningful stabilization and partial recovery of function in ALS. In particular, the observed improvements in bulbar and fine motor domains are noteworthy, as these typically deteriorate progressively in untreated ALS. While the respiratory and gross motor domains remained unchanged, the absence of decline itself may represent a therapeutic benefit, given the expected natural history of the disease. This case demonstrates a modest and safe, but measurable functional improvement on the ALSFRS-R following EV therapy in a patient with ALS. The preservation of motor and respiratory function, combined with improvements in bulbar and fine motor skills, supports the potential role of EVs in modifying disease progression. Further studies are warranted to validate these findings and to define the magnitude, durability, and mechanistic basis of the therapeutic effect.
Discussion
This case report highlights the potential of intravenous administration of human bone marrow-derived extracellular vesicles (EVs) as a novel therapeutic approach in amyotrophic lateral sclerosis (ALS). The patient, an 81-year-old male with a two-year history of ALS and significant neuromuscular decline, underwent a five-month treatment protocol involving monthly EV infusions. During this time, the therapy was well-tolerated, with no reported adverse events or complications. While the intervention was not curative, clinical observations suggest a period of relative functional stability, which is notable given the expected progressive trajectory of ALS at this disease stage.
ALS is marked by continuous motor neuron degeneration leading to increasing muscle weakness, loss of mobility, and respiratory insufficiency. Once respiratory function drops below critical thresholds, such as a forced vital capacity (FVC) below 50–60% predicted, the disease typically progresses more rapidly. In this case, the patient’s FVC was 52% at baseline, and he required non-invasive ventilation (NIV) for up to 9 hours per day. Despite these indicators of advanced disease, the patient remained ambulatory for short distances, retained oral intake, and maintained a stable neurologic baseline throughout the treatment period. These findings may suggest a temporary slowing of disease progression, although natural variability in ALS trajectories and placebo effects cannot be ruled out.
The rationale for using EVs in neurodegenerative conditions is grounded in their ability to modulate inflammation, support neuronal survival, and promote tissue repair through paracrine signaling mechanisms. EVs are known to carry bioactive molecules, including proteins, lipids, and nucleic acids, which can influence cellular processes in recipient cells. Specifically, bone marrow-derived EVs have shown neuroprotective effects in preclinical studies, including models of ALS, through anti-inflammatory, anti-apoptotic, and immunomodulatory pathways. However, the translation of these findings to human subjects remains in its infancy, and limited clinical data support their efficacy in ALS. The therapeutic rationale for EV administration in ALS stems from growing evidence that EVs can influence several key mechanisms of neurodegeneration. ALS pathogenesis involves a complex interplay of glutamate excitotoxicity, mitochondrial dysfunction, oxidative stress, impaired RNA metabolism, and neuroinflammation. In particular, chronic neuroinflammation-driven by activated microglia, astrocytes, and peripheral immune cells—has been recognized as a central feature of disease progression. Bone marrow-derived EVs have been shown to carry immunomodulatory cargo capable of suppressing proinflammatory cytokine expression and promoting a shift toward anti-inflammatory phenotypes in both microglia and T cells.
Furthermore, EVs may play a role in restoring homeostatic communication between neurons and glial cells. Recent studies demonstrate that EVs derived from mesenchymal stem cells (MSCs) can deliver neuroprotective microRNAs (e.g., miR-124, miR-133b, and miR-21) and proteins that support axonal outgrowth, synaptic plasticity, and neuronal survival. In ALS models, MSC-EVs have been shown to reduce oxidative stress and inhibit apoptosis in motor neurons by modulating mitochondrial function and activating anti-apoptotic signaling pathways, such as PI3K/Akt and ERK.
Importantly, this case contributes to the growing body of clinical literature on EV therapy in ALS. To date, only a few published reports have documented the use of EVs in human neurodegenerative diseases. This case provides preliminary evidence that repeated intravenous administration of bone marrow-derived EVs is safe and feasible, even in older adults with multiple comorbidities. The absence of adverse reactions in this patient is encouraging and supports further safety investigations in broader patient populations.
Another hallmark of ALS is the accumulation of misfolded proteins, such as TDP-43, FUS, and SOD1, which contribute to cellular toxicity and motor neuron death. EVs have been implicated both in the spread and clearance of these protein aggregates. Although some reports suggest that EVs may contribute to prion-like propagation of toxic proteins, therapeutic EVs—especially those engineered or selected from anti-inflammatory MSC populations—appear to enhance autophagy and proteasomal degradation pathways, facilitating the removal of misfolded proteins. This dual role underscores the importance of EV source, purification method, and cargo characterization in therapeutic applications.
Notably, preclinical models of ALS have demonstrated functional benefits following EV administration. In SOD1-G93A transgenic mice, intrathecal or intravenous delivery of MSC-derived EVs has led to reduced neuroinflammation, delayed disease onset, and extended survival. These outcomes are consistent with the clinical observations in our patient, whose progression stabilized during the treatment period. While animal models cannot fully recapitulate human ALS, they provide mechanistic insight into how EVs may confer neuroprotection and modulate disease course.
Nonetheless, several limitations must be acknowledged. This is a single, uncontrolled case report, and therefore, no conclusions regarding efficacy can be drawn. The clinical stability observed may be attributed to numerous factors, including ongoing physical therapy, optimal medical management, placebo response, or the natural variability in ALS progression. Additionally, objective outcome measures—such as serial pulmonary function tests, ALS Functional Rating Scale-Revised (ALSFRS-R) scores, or neuroimaging—were not included in this report and would strengthen future studies. Despite the promising biological plausibility and favorable safety profile observed in this case, several limitations must be acknowledged. This is a single, uncontrolled case report, and therefore, no definitive conclusions regarding efficacy can be drawn. Moreover, ALS progression can vary considerably among individuals, and transient stabilization may reflect natural variability rather than treatment effect. Future studies must incorporate standardized outcome measures, including serial ALS Functional Rating Scale-Revised (ALSFRS-R) assessments, quantitative respiratory testing, neuroimaging, and molecular biomarkers to robustly evaluate therapeutic impact.
Conclusion
This case illustrates the feasibility and tolerability of intravenous bone marrow-derived EV therapy in a patient with advanced ALS. While no definitive conclusions regarding efficacy can be drawn from a single case, the findings support further exploration of EV-based interventions in ALS through larger, controlled clinical trials. In conclusion, this case contributes to the growing interest in EV-based therapeutics for neurodegenerative diseases. Given their ability to modulate inflammation, protect neurons, and support tissue repair, EVs represent a promising avenue for ALS treatment. However, their clinical translation requires rigorous investigation through well-controlled trials, detailed characterization of EV cargo, and long-term safety monitoring.
IRB and FDA IND
An Individual Patient Expanded Access Investigational New Drug Application (IND) (Title 21, Code of Federal Regulations (CFR) Part 312) was requested and approved before initiating patient treatment.
WCG IRB
1019 39th Avenue SE, Suite 120 | Puyallup, WA 98374-2115
IRB Tracking Number: 20231989
Sponsor Protocol ID: IND 29481.0
Conflict of Interest
The authors do not have any conflicts of interest in the preparation of this manuscript.
References
- Masrori P, Van Damme P. Amyotrophic lateral sclerosis: a clinical review. Eur J Neurol. 2020;27(10):1918-1929. https://doi:10.1111/ene.14393
- Mehta P, Raymond J, Zhang Y, et al. Prevalence of amyotrophic lateral sclerosis in the United States, 2018. Amyotroph Lateral Scler Frontotemporal Degener. Published online August 21, 2023. https://doi:10.1080/21678421.2023.2245858
- Raghav A, Singh M, Jeong GB, et al. Extracellular vesicles in neurodegenerative diseases: A systematic review. Front Mol Neurosci. 2022;15:1061076. Published 2022 Nov 24. https://doi:10.3389/fnmol.2022.1061076
- Yari H, Mikhailova MV, Mardasi M, et al. Emerging role of mesenchymal stromal cells (MSCs)-derived exosomes in neurodegeneration-associated conditions: a groundbreaking cell-free approach. Stem Cell Res Ther. 2022;13(1):423. Published 2022 Aug 19. https://doi:10.1186/s13287-022-03122-5
- Quan, J., Liu, Q., Li, P. et al. Mesenchymal stem cell exosome therapy: current research status in the treatment of neurodegenerative diseases and the possibility of reversing normal brain aging. Stem Cell Res Ther 16, 76 (2025). https://doi.org/10.1186/s13287-025-04160-5
- van Es MA, Hardiman O, Chio A, et al. Amyotrophic lateral sclerosis. Lancet. 2017;390(10107):2084-2098. https://doi.org/10.1016/S0140-6736(17)31287-4
- Hardiman O, Al-Chalabi A, Chio A, et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers. 2017;3(1):17071. https://doi.org/10.1038/nrdp.2017.71
- Bourke SC, Tomlinson M, Williams TL, Bullock RE, Shaw PJ, Gibson GJ. Effects of non-invasive ventilation on survival and quality of life in patients with amyotrophic lateral sclerosis: a randomized controlled trial. Lancet Neurol. 2006;5(2):140–147. https://doi.org/10.1016/S1474-4422(05)70326-4
- Chiò A, Logroscino G, Traynor BJ, et al. Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology. 2013;41(2):118–130. https://doi.org/10.1159/000351153
- Colombo M, Raposo G, Théry C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. 2014;30:255–289. https://doi.org/10.1146/annurev-cellbio-101512-122326
- Théry C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2(8):569–579. https://doi.org/10.1038/nri855
- Yáñez-Mó M, Siljander PR-M, Andreu Z, et al. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles. 2015;4(1):27066. https://doi.org/10.3402/jev.v4.27066
- Bonafede R, Mariotti R. Exosomes and their role in amyotrophic lateral sclerosis. Neurobiol Dis. 2017;103:124–131. https://doi.org/10.1016/j.nbd.2017.04.011
- Soria FN, Pampliega O, Bourdenx M, et al. Exosomes, an unmasked culprit in neurodegeneration? J Neurosci. 2017;37(37):9240–9247. https://doi.org/10.1523/JNEUROSCI.0741-17.2017
- Taylor JP, Brown RH Jr, Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016;539(7628):197–206. https://doi.org/10.1038/nature20413
- Bruno S, et al. Mesenchymal stem cell-derived EVs protect against acute kidney injury. J Am Soc Nephrol. 2009;20(5):1053–1067. https://doi.org/10.1681/ASN.2008070798
- Di Trapani M, et al. Differential and transferable modulatory effects of mesenchymal stromal cell-derived extracellular vesicles on T, B and NK cell functions. Sci Rep. 2016;6:24120. https://doi.org/10.1038/srep24120
- Xin H, et al. MicroRNA cluster miR-17-92 enriched exosomes promote neuroplasticity and functional recovery after stroke in rats. Stroke. 2017;48(3):747–753. https://doi.org/10.1161/STROKEAHA.116.015204
- Lee M, et al. Mesenchymal stem cell-derived small extracellular vesicles preserve hippocampal neuronal density and function in a mouse model of Alzheimer’s disease. Sci Rep. 2018;8:10713. https://doi.org/10.1038/s41598-018-29131-0
- Bonafede R, et al. Exosome-derived microRNAs from mesenchymal stem cells: therapeutic perspectives in neurodegenerative diseases. Aging (Albany NY). 2016;8(9):2270–2291. https://doi.org/10.1083/jcb.201504057
- Leung C, Tan SH, Barker RA. The role of extracellular vesicles in the pathophysiology of neurodegenerative diseases and their therapeutic potential. Cell Transplant. 2021;30:1–15. https://doi.org/10.1177/09636897211025262
- Lener T, Gimona M, Aigner L, et al. Applying extracellular vesicles-based therapeutics in clinical trials – an ISEV position paper. J Extracell Vesicles. 2015;4(1):30087. https://doi.org/10.3402/jev.v4.30087
- Feiler MS, et al. TDP-43 is intercellularly transmitted across axon terminals. J Cell Biol. 2015;211(4):897–911. https://doi.org/10.1083/jcb.201504057
- Guo BB, et al. Therapeutic potential of human adipose-derived extracellular vesicles in amyotrophic lateral sclerosis. Front Neurosci. 2020;14:581689. https://doi.org/10.3389/fnins.2020.581689
- Paganoni S, Macklin EA, Lee A, et al. Clinical trial outcomes and placebo effects in amyotrophic lateral sclerosis. Neurology. 2019;93(4):e358–e367. https://doi.org/10.1212/WNL.0000000000007856
- Chio A, Hammond ER, Mora G, Bonito V, Filippini G. Prognostic factors in ALS: A critical review. Amyotroph Lateral Scler. 2009;10(5-6):310–323. https://doi.org/10.3109/17482960802566824
- Crose, Joshua J, Arezou Crose, John T Ransom, and Amy L Lightner. 2024. “Bone Marrow Mesenchymal Stem Cell-Derived Extracellular Vesicle Infusion for Amyotrophic Lateral Sclerosis.” Neurodegenerative Disease Management 14 (3–4): 111–17. doi:10.1080/17582024.2024.2344396.