

Clinical Insights on Celiac Disease and Related Disorders
A Clinical Review of Collagenous Sprue, Refractory Celiac Disease, Ulcerative Jejunitis, and Enteropathy-Associated T-cell Lymphoma
Cesar Moreno, MD¹,², Hunter Chance Dickson, DO¹,², Gannon Ray, MD¹,³, Benjamin Niland, MD¹,²
- University of South Alabama, Department of Internal Medicine, Mobile, AL
- University of South Alabama, Division of Gastroenterology, Mobile, AL
- University of South Alabama, Department of Hematology and Medical Oncology, Mobile, AL
# These authors contributed equally to this work
OPEN ACCESS
PUBLISHED: 31 December 2025
CITATION: Moreno, C., Dickson, HC., et al., 2025. A Clinical Review of Collagenous Sprue, Refractory Celiac Disease, Ulcerative Jejunitis, and Enteropathy-Associated T-cell Lymphoma. Medical Research Archives, [online] 13(12). https://doi.org/10.18103/mra.v13i12.7053
COPYRIGHT: © 2025 European Society of Medicine. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
DOI https://doi.org/10.18103/mra.v13i12.7053
ISSN 2375-1924
ABSTRACT
Celiac disease is a chronic immune-mediated enteropathy that affects approximately 1% of the global population. Most patients respond well to a strict gluten-free diet; however, a small subset develop worsening or life-threatening symptoms despite gluten avoidance. These cases present diagnostic and therapeutic challenges and raise concern for alternative or overlapping disorders. Collagenous sprue, refractory celiac disease types I and II, ulcerative jejunitis, and enteropathy-associated T-cell lymphoma are all types of celiac-related disorders that present in patients with nonresponsive celiac disease. Collagenous sprue is defined by a thickened subepithelial collagen band and celiac-like histologic features of the small intestine. It remains uncertain whether collagenous sprue lies within the celiac disease spectrum or represents a distinct entity. Refractory celiac disease is a rare form of celiac disease unresponsive to a gluten-free diet and is subdivided into types I and II. Type II is defined by a clonal population of aberrant intraepithelial lymphocytes that accumulate lymphomagenic mutations. This premalignant condition may present with ulcerative jejunitis or progress to enteropathy-associated T-cell lymphoma, an aggressive intestinal lymphoma with poor prognosis. Diagnosis of refractory celiac disease is complex and requires immunohistochemistry, flow cytometry, and T-cell receptor clonality testing. Treatment may include immunosuppressants, chemotherapy, and stem cell transplantation. This review summarizes current knowledge of collagenous sprue, refractory celiac disease, ulcerative jejunitis, and enteropathy-associated T-cell lymphoma. It provides a unified framework that outlines presentation, pathogenesis, diagnosis, and treatment. The goal is to guide clinicians toward earlier disease recognition and timely management.
Keywords
Celiac disease, collagenous sprue, refractory celiac disease, ulcerative jejunitis, enteropathy-associated T-cell lymphoma
Introduction
Celiac disease (CD) has ancient roots dating back to Aretaeus of Cappadocia, a Greek physician around 250 AD, who first described a syndrome of abdominal pain, chronic diarrhea, and malnutrition. Since that time, our understanding of celiac disease has evolved to include the identification of gluten as the trigger of immune-mediated bowel injury leading to a wide spectrum of disease manifestation. The condition occurs in specific individuals who carry a genetic predisposition. Elimination of gluten from the diet is the primary treatment, which leads to resolution of both symptoms and histologic derangements in the vast majority of patients. There is a subset of patients who experience persistent symptoms and mucosal injury despite adherence to a gluten-free diet (GFD). From this group of patients, approximately 10% have refractory celiac disease (RCD), which can progress to ulcerative jejunitis (UJ) and enteropathy-associated T-cell lymphoma (EATL). There are other disorders such as collagenous sprue (CS) that share overlapping features with CD; nevertheless, it remains unclear whether collagenous sprue represents a distinct disease or exists within the spectrum of CD. This review aims to explore the clinical and pathologic spectrum of celiac disease and sprue-like enteropathies, including collagenous sprue, refractory celiac disease, ulcerative jejunitis, and enteropathy-associated T-cell lymphoma.
Celiac disease
One of the first modern descriptions of celiac disease came from Dr. Samuel Gee in 1888 when he reported symptoms of diarrhea and malabsorption in children and hypothesized that the etiology was linked to diet. Fifty years later, the Dutch pediatrician, Dr. Willem Karel Dicke, described the link between gluten and celiac disease. He observed that his hospitalized patients improved during World War II when breads and cereals were scarce. He showed that a gluten-free diet improved symptoms of celiac disease.
Gluten (“glue” in Latin) is found in rye, barley, and wheat and is made up of gliadins and glutenins, which form a glue-like substance when mixed with water. Large gluten peptides reach the small bowel largely undigested due to their high proline and glutamine content, which resist enzyme breakdown.
PATHOPHYSIOLOGY
Gluten peptides interact with innate immune cells and trigger the release of interleukin-15 (IL-15), which promotes activation of intraepithelial lymphocytes (IELs). The cytotoxic activity of IELs against enterocytes causes epithelial cell damage. In individuals with human leukocyte antigen (HLA) DQ2 or DQ8 alleles, gluten undergoes deamidation by tissue transglutaminase 2 (tTG2), thus increasing its affinity for HLA-DQ2/DQ8 proteins on antigen-presenting cells. This process enhances activation of gluten-specific CD4+ T helper cells in the lamina propria. These T helper cells produce pro-inflammatory cytokines and promote B cell activation. In turn, the gluten-specific B cells produce antibodies such as anti-tTG2, anti-deamidated gliadin peptide, and anti-endomysial antibodies that contribute to tissue damage.
PRESENTATION AND EPIDEMIOLOGY
Patients with CD can present with chronic diarrhea, weight loss, bloating, abdominal pain, anemia, and nutrient deficiencies. A systematic review and meta-analysis by Singh et al estimated a global prevalence of celiac disease of 1.4% based on serology and 0.7% based on biopsy. The prevalence of CD is higher in populations with European ancestry due to increased incidence of HLA-DQ2 and HLA-DQ8 haplotypes. Prevalence is up to two times higher in women compared to men and increased in patients with autoimmune conditions. Additionally, prevalence is increasing in developing countries due to dietary westernization and greater disease recognition.
DIAGNOSIS
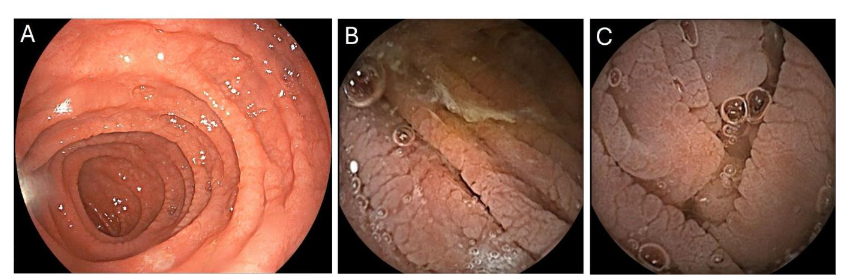
Celiac disease can be diagnosed using symptomatology, serologic markers including elevated tissue transglutaminase IgA, and small bowel biopsies showing villous atrophy, crypt hyperplasia and increased IELs. HLA-DQ2 and/or DQ8 testing is useful due to its high negative predictive value which can exclude CD in cases with discordant serology and histology findings or for those patients already on GFD without antecedent diagnostic testing. Endoscopic evaluation can reveal non-specific features suggestive of villous atrophy such as flattening of folds, scalloping, nodularity and fissuring.
TREATMENT
A strict gluten-free diet remains the only effective and recommended therapy for celiac disease. Maintaining a strict GFD and avoiding inadvertent exposure are challenging and leave a therapeutic window open to novel therapies. There are several drugs under investigation that target different steps in the CD pathogenesis including gluten degradation, intestinal permeability, immunomodulation and gluten tolerance induction. Notably, ZED-1227, an oral transglutaminase 2 inhibitor, has shown reduction in mucosal injury and is expected to progress in clinical evaluation. Novel agents in various stages of investigation may eventually change the landscape of CD treatment. Presently, a strict GFD is the only therapy approved for routine CD management. Medications for the management of refractory celiac disease will be discussed later in this review.
Collagenous sprue
Collagenous sprue is characterized by the presence of malabsorptive symptoms and a subepithelial collagen band.
PREVALENCE AND EPIDEMIOLOGY
CS is a rare disorder with an unknown prevalence. There are studies describing cohorts from 1 to 35 patients with collagenous sprue spanning decades. The youngest patient with CS was a 3-month-old infant in Japan successfully treated with steroids and immunomodulator therapy. CS predominantly affects older adults (60-70s years) with female predominance (approximately 2 to 1 female:male ratio). Rubio-Tapia et al described 30 patients with CS undergoing GFD and steroid treatment, 29 of which were White and 1 was Black. This race disparity may represent underdiagnosis in other patient populations rather than true race-disease association.
PATHOGENESIS
The mechanism of CS remains unknown. CS may be its own distinct pathological entity or lie within the celiac disease spectrum. The inconsistent presence of celiac serologies and variable HLA-DQ2/DQ8 association suggests that collagenous sprue may be a distinct entity outside of the celiac disease spectrum. Additionally, CS-like histology can be seen with exposure to medications such as olmesartan, nonsteroidal anti-inflammatory drugs, proton pump inhibitors and statins which favors a non-gluten mediated mechanism. Finally, some patients with CS may not respond to GFD and require immunosuppression or withdrawal of offending medications which differs from the treatment of uncomplicated CD. Evidence that CS may lie within the CD spectrum includes shared HLA DQ2/DQ8 genotypes and diagnosis of CD in several patients with CS. Additionally, some patients with CS can respond to GFD which suggests a gluten sensitivity mechanism. CS and CD share similar histologic features, including villous atrophy and intraepithelial lymphocytosis. Notably, subepithelial collagenous thickening can be present in certain cases of CD.
Although the mechanism of CS is not fully understood, it likely involves immune-mediated injury triggered by immune dysregulation or environmental factors. CS coexists with other autoimmune disorders, which points to a possible link between immune dysregulation and CS. Rubio et al reported 70% of patients with CS had immune-mediated disorders such as CD (most common), microscopic colitis, autoimmune thyroiditis, and hepatitis. Similarly, Vakiani et al reported 63% of patients with CS had autoimmune or immune disorders. Notably, authors suggested that the degree of collagen deposition may be the result of the extent of immune dysregulation, as patients with severe fibrosis had more autoimmune conditions. An increase in IgG4-positive plasma cells found in a subset of patients with CS adds to the theory that autoimmune disease plays a role in the pathogenesis of CS. There may be a shared mechanism between CS and other collagenous gastrointestinal diseases such as collagenous gastritis and collagenous colitis given similar histologic appearance and associated medication triggers. A similar association between CD, especially refractory CD, with other collagenous disorders such as microscopic colitis is well established.
PRESENTATION AND DIAGNOSIS
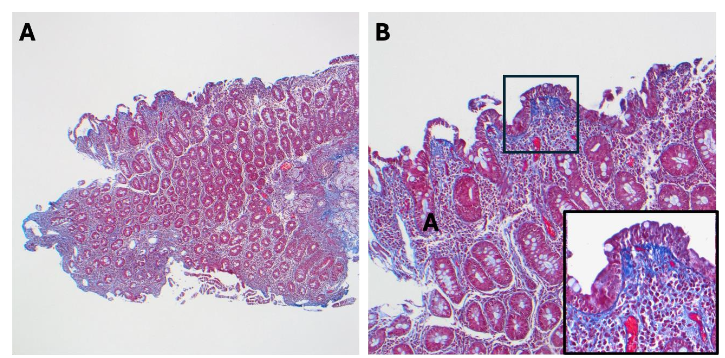
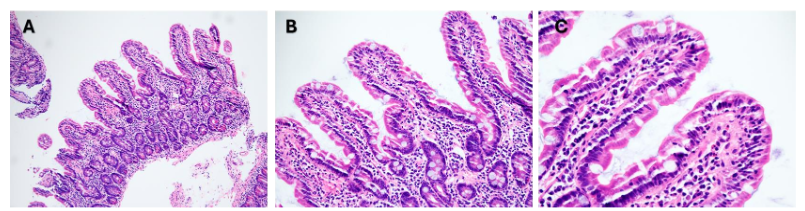
There are no formal guidelines for the diagnosis of CS. The final diagnosis can be made taking into account the clinical presentation and histology while excluding other etiologies. The clinical presentation most commonly associated with CS includes chronic diarrhea, weight loss, malnutrition, and abdominal pain, which may be refractory to GFD. The definitive diagnosis of CS requires small bowel biopsies. The hallmark histologic feature is a thick subepithelial collagen band (greater than 10 μm), while other features such as villous atrophy and IELs may be present. Additional evaluation to exclude other causes of enteropathy, such as medications and autoimmune enteropathy, should be pursued.
TREATMENT
CS was initially thought to have a poor prognosis due to progressive malabsorption, severe clinical presentation, hospitalization, frequent total parenteral nutrition requirements, and lack of effective treatments at the time; however, more studies have shown favorable outcomes. GFD may prove effective in CS management, especially in patients with concomitant CD. Nevertheless, when compared to CD, the response to GFD in CS is low and ranges from 0 to 21% across several studies. In those who do not respond to GFD, corticosteroid therapy should be initiated. In a study of 30 patients with CS, 87% received corticosteroids, and 80% achieved a clinical response. Budesonide was the most commonly used corticosteroid, likely due to its favorable safety profile. Notably, budesonide is used as first-line management in refractory celiac disease and collagenous colitis. In steroid-refractory cases, immunomodulators such as azathioprine, 6-mercaptopurine and thioguanine have proven helpful to various degrees. Immunomodulators may be used alongside corticosteroids. Medication withdrawal in cases of suspected drug-induced CS should be considered. Possible culprit medications include olmesartan, non-steroidal anti-inflammatory drugs, proton pump inhibitors, and statins.
Nonresponsive celiac disease
Nonresponsive celiac disease (NRCD) is defined as the persistence or recurrence of symptoms, signs, and laboratory changes characteristic of celiac disease despite 6 to 12 months of gluten-free diet. The estimated prevalence of NRCD in patients with CD is 22%. The most common cause is inadvertent gluten ingestion which occurs in 33-50% of cases of NRCD. The 2019 European Society for the Study of Coeliac Disease (ESsCD) guideline discourages the term NRCD as most of the patients in this category are considered GFD slow-responders or may have an alternative diagnosis for their symptoms. Nonetheless, the ESsCD guideline regarding management of GFD slow-responders is similar to the American College of Gastroenterology guidelines. The evaluation of NRCD should start with confirmation of celiac disease by reviewing serologies and histology. After the diagnosis is confirmed, inadvertent gluten exposure should be evaluated with celiac serologies and consultation with an expert dietitian. An objective assessment of inadvertent gluten exposure in NRCD with urine or stool gluten immunogenic peptides (GIP) is recommended.
If gluten exposure is excluded and symptoms persist, repeat duodenal biopsies should be obtained to assess for ongoing villous atrophy. If villous atrophy is identified then consider CD with slow mucosal healing beyond the generally accepted 6-12 month GFD treatment time frame. Additionally, non-CD etiologies of villous atrophy such as autoimmune enteropathy, small intestinal bacterial overgrowth, tropical sprue, collagenous sprue, eosinophilic enteritis, common variable immunodeficiency, drug-induced enteropathy and Crohn’s disease should be ruled out. If there is no villous atrophy on repeat duodenal biopsies despite persistent symptoms then other disorders that may coexist or mimic CD such as irritable bowel syndrome, lactose intolerance, fructose intolerance, microscopic colitis and pancreatic insufficiency should be considered. Colon biopsies should be considered in patients without villous atrophy and persistent diarrhea to rule out microscopic colitis and inflammatory bowel disease. Finally, if other causes of villous atrophy have been excluded then an evaluation for refractory celiac disease should be performed.
Refractory Celiac Disease
Refractory celiac disease (RCD) refers to persistent symptoms and villous atrophy in patients with confirmed CD despite GFD for 12 months or longer, after excluding other possible causes of NRCD. There are 2 subtypes of RCD based on immunophenotypic and molecular characteristics of IELs. RCD type I (RCD I) is characterized by normal polyclonal IELs while RCD type II (RCD II) has a clonal population of aberrant IELs. RCD is rare with a prevalence of approximately 1% in patients with CD and up to 23% in patients with NRCD. The reported prevalence of RCD is decreasing in newer studies which may be due to improved differentiation from conditions that mimic RCD, improved gluten detection, and reduced referral center bias.
Refractory Celiac Disease Type I
RCD I is the more common and less aggressive RCD subtype. It accounts for approximately 75% of all RCD cases and it typically affects older individuals with a higher prevalence in females. RCD I has a more favorable prognosis with a 5-year survival of 80% to 90%.
PATHOGENESIS AND DIAGNOSIS
RCD I is defined by polyclonal IELs that are phenotypically normal (expressing surface CD3 and surface CD8 markers). The pathogenesis of RCD I is incompletely understood. A possible mechanism consists of enhanced immune response to trace amounts of gluten in genetically predisposed individuals such as those with HLA DQ2.5 homozygosity. Another possible mechanism involves IL-15 overexpression independent of gluten exposure. IL-15 leads to activation of dysregulated IELs with improved survival and independence from regulatory T cells which contributes to chronic mucosal damage. Another trigger for IL-15 upregulation includes type 1 interferons which can be seen in the antiviral response. Additionally, IL-15 overexpression may represent an autoimmune process supported by the increased autoimmune disease burden and positive response to immunosuppressive therapy observed in patients with RCD I.
Diagnosis of RCD I involves the use of immunohistochemistry, flow cytometry and polymerase chain reaction (PCR)-based T-cell receptor molecular studies to look for polyclonal, phenotypically normal IELs characteristic of RCD I. IELs in RCD I have a normal surface marker profile which includes CD3+, CD8+, T-cell receptor (TCR). Additionally, PCR testing shows diversity of T-cell receptor gamma (γ) and beta (β) proteins which indicates polyclonal IELs and excludes clonal dominance.
TREATMENT
Treatment goals include symptom improvement, mucosal healing, improvement in nutritional status and prevention of progression to RCD II or EATL. The foundation of care remains strict GFD with regular follow up with a multidisciplinary team to monitor nutritional status. First-line therapy consists of corticosteroids. Open-capsule budesonide (OCB) is the preferred choice due to low systemic absorption and improved delivery to the small bowel. OCB should be given for at least 3 months. In a retrospective cohort study of 43 patients with RCD I and 13 patients with RCD II, Mukewar et al showed that OCB achieved complete and partial clinical improvement in 92% of patients and histologic improvement in 89% of patients despite prior treatment failure with regular budesonide, systemic corticosteroids and azathioprine in 51% of the cohort. OCB was well tolerated with only three patients experiencing adverse effects (edema, fatigue and nausea). Nearly half of the patients were successfully tapered off OCB while the rest stayed at lower doses. The median treatment duration was 15 months. OCB was dosed 3 mg three times daily with each dose swallowed in such a way as to target the upper small bowel (open-capsule, crushed in applesauce), mid small bowel (open-capsule in applesauce) and distal small bowel (intact capsule).
If OCB is unavailable or if patients have severe or rapidly progressive symptoms then prednisone 40 – 60 mg daily with gradual taper can be used. Second-line therapy consists of thiopurines. Azathioprine (2-2.5 mg/kg/day) is the preferred thiopurine and can be used for patients with partial response to corticosteroids, relapse during steroid taper, or for those who have become steroid dependent. Duodenal biopsy should be performed to assess response to azathioprine after 3 months of therapy. Lack of response should prompt optimization of thiopurine dose and reevaluation of alternative diagnosis to RCD. Annual endoscopy with biopsy should be performed to evaluate for transformation to RCD II.
Refractory Celiac Disease Type II
RCD II is an extremely rare and severe form of refractory celiac disease. RCD II has a worse prognosis than RCD I with a 5-year survival of 44-58% driven by the high risk of progression to EATL. Al-Toma et al found that 26 of 50 patients (52%) developed secondary EATL within 4-6 years. Clinical presentation of patients with RCD includes malabsorptive symptoms and laboratory abnormalities such as diarrhea, weight loss, abdominal pain, fatigue, hypoalbuminemia, anemia that are generally worse in patients with RCD II. The median age at diagnosis is slightly older in RCD II compared to RCD I. RCD patients are predominantly females with less pronounced female representation in RCD II.
PATHOGENESIS AND DIAGNOSIS
The key feature of RCD II is the clonal expansion of aberrant IELs. Aberrant IELs may originate from immune lymphoid cell (ILC)-like precursors whose differentiation stopped at the pre-natural killer (NK) cell or pre-T cell stages rather than mature T cells who have lost their typical markers. The arrested development of aberrant IELs increases their susceptibility to clonal expansion and mutations which is enhanced by the IL-15 rich environment of CD. One such mutation involves the Janus kinase (JAK)1/signal transducer and activator of transcription (STAT) 3 signaling pathway that leads to increased proliferation, decreased apoptosis and reduced response to regulatory T cells in response to several cytokines including IL-15.
Aberrant IELs have an abnormal immunophenotype that lacks surface CD3, CD8 and has cytoplasmic CD3. Flow cytometry can distinguish surface from cytosolic CD3 and should be used over IHC if available due to its greater sensitivity and specificity. The aberrant IEL threshold for RCD II diagnosis is > 50% of the total IEL population by IHC or ≥ 20% by flow cytometry. IHC or flow cytometry should be followed by TCR clonal analysis to enhance diagnostic accuracy. IEL clonality alone should not be used to establish the diagnosis of RCD II as clones can occur in uncomplicated CD and RCD II. The diagnosis of RCD II should be made by taking into consideration the clinical presentation, histology, IEL immunophenotype and clonality.
Singh et al used multi-omic single-cell analysis in patients with RCD to show the presence of several lymphoma driver somatic mutations, especially in RCD II, and proposed a new model to classify celiac disease on a continuum based on accumulation of mutated IELs. Using this continuum model, no mutated IELs corresponds to CD in remission and a markedly expanded mutation-rich clonal population corresponds to RCD II.
TREATMENT
The evidence that guides RCD II treatment is limited. Presently, there is no therapy that decreases aberrant IELs and prevents RCD II to EATL progression. After initial RCD II diagnosis, UJ and EATL should be excluded as these conditions can alter management. Imaging with capsule endoscopy and adjunctive computed tomography (CT) or magnetic resonance (MR) enterography can be used to evaluate for UJ and EATL. The treatment goals and initial treatment of RCD II mirrors that of RCD I as described in the RCD I section above. Treatment should start with strict GFD, nutritional support and OCB.
Despite initial improvement in symptoms and histology while on OCB, patients may become refractory to corticosteroids or develop EATL. If a patient is not improving while on corticosteroids, clinicians should consider evaluation at a center with RCD experience or enrollment in clinical trials. Additionally, patients should have regular follow up with a multidisciplinary team that includes gastroenterologists and dietitians.
For patients with life-threatening disease and who have had partial or no response to corticosteroids, limited therapies exist. These may include purine analogs such as cladribine, small bowel mesalamine release, biologics, autologous stem cell transplantation, anti IL-15 monoclonal antibodies and tofacitinib. AMG 714, an anti-IL-15 monoclonal antibody, improved symptoms and stabilized TCR clonality in patients without reduction in aberrant IELs. Cladribine, a cytotoxic purine nucleoside analog, targets abnormal lymphoid cells with high or low proliferative activity. Cladribine showed clinical response in 81% of patients and aberrant IEL reduction in 41% of patients with an improved 5-year survival in the responders compared to the non-responders. Progression to EATL was observed in 16% of patients. ASCT showed clinical and histologic improvement in 13 patients who failed cladribine with a 4-year survival of 66% in the post-transplant group. There was no decrease in aberrant IELs which suggests this metric may not be a reliable marker of treatment response or prognosis. Tofacitinib, a JAK1/3 inhibitor, was used to treat 6 patients with RCD II. While treatment did not lead to reduction in aberrant IELs, there was rapid symptom response and histologic improvement which may be due to aberrant IEL function suppression. There is a need for additional studies to develop standardized treatments that achieve clinical response, reduce aberrant IELs and decrease progression to EATL.
Ulcerative Jejunitis in Refractory Celiac Disease
INTRODUCTION
Ulcerative jejunitis is a rare and serious complication of CD, most commonly found in RCD II. Although most reported cases of UJ are associated with RCD II, UJ is still exceedingly rare. In a study of 713 patients with CD, only 5 patients developed UJ. Risk factors include older age at diagnosis, noncompliance to a strict GFD, persistent villous atrophy and severe malabsorption.
PATHOPHYSIOLOGY
UJ is characterized by chronic intestinal inflammation despite adherence to a GFD. Aberrant IELs in RCD II release cytokines such as IL-15 and IFN-γ leading to chronic mucosal inflammation and villous atrophy. This persistent breakdown of mucosa within the small intestine can result in ulcer formation, most commonly in the proximal jejunum. Other manifestations have been reported such as transverse circumferential ulcerations, stricture formation, and fissuring type ulcerations. The accumulation of these clonal aberrant IELs may represent an early step in lymphomagenesis, and UJ is considered a precursor to EATL.
PRESENTATION AND DIAGNOSIS
Patients may present with nonspecific symptoms such as abdominal pain, chronic diarrhea, weight-loss, and malabsorption. They may also present with acute symptoms such as gastrointestinal obstruction, bleeding, perforation, and peritonitis. UJ should be suspected in any patient with RCD II who fails to improve following a strict GFD and aggressive nutritional support as these symptoms overlap with other gastrointestinal disorders and can make the diagnosis challenging.
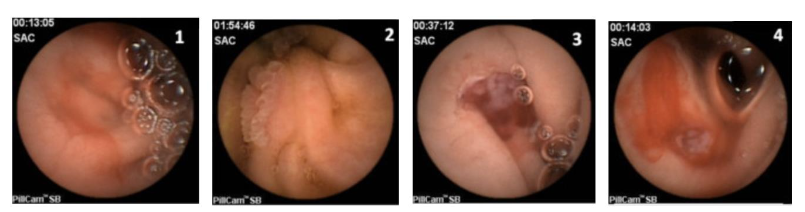
Due to the location of UJ, traditional endoscopy often fails to detect the disease. Consequently, advanced imaging techniques such as video capsule endoscopy (VCE) and double-balloon endoscopy (DBE) are often utilized. DBE allows for direct biopsy and is considered the most effective tool for diagnosing UJ. VCE is a less invasive method that can aid in visualizing the extent of the disease as long as strictures have been ruled out.
PATHOLOGY
Considering that UJ is highly associated with RCD II, the histology of ulcerations typically reveals villous atrophy, crypt distortion, and infiltration of aberrant IELs, as seen in RCD II. These aberrant IELs lack typical surface CD3, CD4, CD8, and TCR but retain cytoplasmic CD3 and express surface CD103.
TREATMENT
There are no standardized guidelines for the treatment of UJ. Adherence to a strict GFD and nutritional support are essential. In one case involving a 9-year-old girl, strict adherence to GFD resulted in normalization of the jejunal mucosa on follow-up endoscopy without the need for immunosuppressive therapy. In other cases, more aggressive therapy may be necessary. In a case involving an adult patient with RCD II and UJ who did not respond to systemic corticosteroids, surgical resection of the affected jejunal segment followed by GFD and OCB resulted in symptom resolution. Targeted immunosuppressive agents, such as JAK inhibitors, have also been utilized in the treatment of UJ. A study evaluating the use of Tofacitinib in patients with RCD II showed two patients with UJ who had clinical and histologic remission while on tofacitinib. These cases highlight the need for an individualized treatment approach for patients with UJ.
Enteropathy Associated T-cell Lymphoma
INTRODUCTION
EATL is a rare and aggressive intestinal T-cell non-Hodgkin lymphoma. EATL is not a single disease but rather two distinct disorders that differ cytologically, genetically, and in their association with CD. Type I EATL is most often diagnosed in patients with refractory or untreated CD and can have either a monomorphic or pleomorphic cytologic appearance. In contrast, type II EATL is commonly characterized by a monomorphic appearance and can be seen in patients with and without a history of CD. Type II EATL has been renamed monomorphic epitheliotropic intestinal T-cell lymphoma (MEITL).
EPIDEMIOLOGY AND RISK FACTORS
EATL is estimated to account for 1-2% of all non-Hodgkin lymphoma cases globally. It has an annual incidence rate of 0.5-1 per million, is more frequently seen in populations with a higher incidence of CD, such as Western countries, and is more commonly seen in males over the age of 60. The disease commonly localizes to the jejunum and ileum. Risk factors for the development of EATL include nonadherence to GFD, late diagnosis of CD, and the HLA-DQ2 haplotype, especially those with HLA-Dq2.5 homozygosity. Characteristic genetic alterations include gain of chromosome 9q31.3 and deletion of 16q12.1. Most reported cases are in patients with a diagnosis of RCD II. In one retrospective study, 52% of patients with RCD II developed EATL within 4 to 6 years.
PATHOPHYSIOLOGY
EATL arises from the malignant transformation of aberrant IELs in RCD II. Aberrant IELs create a chronic inflammatory environment within the intestinal mucosa in which they acquire mutations that lead to unregulated clonal expansion resulting in lymphomagenesis. Patients with a higher population of aberrant IELs are more likely to develop EATL. Key molecular drivers of this malignant transformation include overexpression of IL-15 and mutations in the JAK1/STAT3 pathway. Overexpression of IL-15 promotes IEL survival and the cytotoxic activity by impairing normal regulatory T cell function, while mutations in the JAK1/STAT3 promote resistance to apoptosis.
PRESENTATION AND DIAGNOSIS
Common presenting symptoms of EATL include abdominal pain, diarrhea, fever, night sweats, and unintentional weight loss. In nearly half of patients, the initial presentation is an acute complication such as gastrointestinal bleeding or bowel obstruction and EATL is diagnosed only after surgery. Laboratory abnormalities that should raise suspicion for EATL in a patient with RCD II include hypoalbuminemia, anemia, peripheral eosinophilia, and elevated lactate dehydrogenase.
The overlap between EATL and other inflammatory bowel conditions can make the diagnosis challenging. MR enteroclysis, CT scans and positron emission tomography (PET) can be used; however, the sensitivity of CT scans and PET scans has been limited in detecting early-stage lymphoma making the diagnosis highly reliant on clinical suspicion and other forms of comprehensive testing, such as DBE and VCE, as these can detect lesions that are typically found in the jejunum and ileum.
PATHOLOGY
Histopathologic, immunophenotypic, and molecular techniques are essential for confirming the diagnosis of EATL, although the features of EATL are highly variable. Commonly used methods include IHC, flow cytometry, and TCR gene rearrangement studies. These methods reveal the presence of aberrant IELs, a hallmark of the disease. However, the sensitivity of IHC and TCR rearrangement testing decreases in patients with mild elevations in aberrant IELs. In such cases, flow cytometry should be used. Histologically, small intestinal biopsies in EATL typically reveal pleomorphic lymphoid cells with increased mitotic activity. These tumor cells exhibit a characteristic immunophenotype: CD3+, CD4−, CD5−, CD7+, CD8−, CD56−, CD103+, CD30+, and variably TCRβ+. They also display a cytotoxic profile with expression of perforin, granzyme B, and TIA-1, supporting their origin from intraepithelial T lymphocytes. Surrounding mucosal findings often reflect the underlying CD, with villous atrophy, crypt hyperplasia, and an increase in IELs.
PROGNOSIS AND TREATMENT
At diagnosis, most patients have either stage III or IV disease. EATL carries a dismal prognosis with a survival of 7.5 months. Roughly 40% of patients treated with chemotherapy achieve complete remission. There is no current standardized treatment for EATL. Surgery is often utilized for local debulking and resecting masses that are at high risk of developing bleeding, obstruction, or perforation. Chemotherapy with CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) is most often used. However, it cannot be administered in over 50% of cases secondary to low performance status and advanced age at time of diagnosis.
Some studies have examined the use of combining brentuximab vedotin with cyclophosphamide, doxorubicin, and prednisone (CHP). In one study, brentuximab vedotin plus CHP was compared to CHOP in patients with CD30+ peripheral T-cell lymphomas, including a small number of patients with EATL. This study demonstrated superior progression-free survival and overall survival in the brentuximab vedotin plus CHP group. In a subsequent study, this same regimen followed by high-dose therapy and autologous stem cell transplantation (HDT-ASCT) was studied in newly diagnosed EATL patients and resulted in a 2-year progression-free survival of 63% and an overall survival of 68%.
Other novel therapies and radiotherapy have also been studied. A monoclonal antibody targeting CD52 on T-lymphocytes, alemtuzumab, has shown varying results when combined with chemotherapy in EATL. Some case reports demonstrated complete responses while other studies have reported relapse or progression. Cladribine has demonstrated clinical and histologic responses in patients with RCD II, potentially reducing progression to EATL. However, its effectiveness in overt EATL remains unclear. Radiotherapy has been used as an adjunct to chemotherapy. Other agents that have been studied include histone deacetylase inhibitors such as romidepsin and belinostat, and experimental therapies targeting IL-15, which contributes to IEL survival and lymphomagenesis.
Conclusion
Celiac disease is a complex, immune-mediated enteropathy of varying severity. It is associated with additional forms of small bowel injury apart from villous atrophy such as collagenous sprue and ulcerative jejunitis. In rare cases, it can progress to an immune-dysregulated refractory form that leads to injury independent of gluten. This includes refractory celiac disease type II which carries a poor prognosis due to its risk of progression to enteropathy-associated T-cell lymphoma. While a strict gluten free diet is fundamental in the management of celiac disease, therapeutic options are limited for patients who develop refractory disease or progress to EATL. Future studies are needed to shed more light on the molecular and genetic mechanisms of RCD and EATL with the objective of finding those at highest risk. Additional research is needed to develop standardized therapies for the management of these patients.
Conflicts of Interest Statement
The authors report no conflicts of interest pertaining to this work.
Funding Statement
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Reference list
- Freeman HJ. The Neolithic revolution and subsequent emergence of the celiac affection. Int J Celiac Dis. 2013;1(1):19-22.
- Dowd B, Walker-Smith J. Samuel Gee, Aretaeus, and the coeliac affection. Br Med J. Apr 6 1974;2(5909):45-7. doi:10.1136/bmj.2.5909.45
- van Berge-Henegouwen GP, Mulder CJ. Pioneer in the gluten free diet: Willem-Karel Dicke 1905-1962, over 50 years of gluten free diet. Gut. 1993;34(11):1473-1475. doi:10.1136/gut.34.11.1473
- Iversen R, Sollid LM. The Immunobiology and Pathogenesis of Celiac Disease. Annu Rev Pathol. Jan 24 2023;18:47-70. doi:10.1146/annurev-pathmechdis-031521-032634
- Lebwohl B, Rubio-Tapia A. Epidemiology, Presentation, and Diagnosis of Celiac Disease. Gastroenterology. 2021-01-01 2021;160(1):63-75. doi:10.1053/j.gastro.2020.06.098
- Singh P, Arora A, Strand TA, et al. Global Prevalence of Celiac Disease: Systematic Review and Meta-analysis. Clin Gastroenterol Hepatol. Jun 2018;16(6):823-836.e2. doi:10.1016/j.cgh.2017.06.037
- Lebwohl B, Green PHR. Celiac Disease. In: Feldman M, ed. Sleisenger and Fordtran’s Gastrointestinal and Liver Disease. Eleventh Edition ed. 2021:1736-1755e6:chap 107.
- Fasano A, Catassi C. Celiac Disease. New England Journal of Medicine. 2012;367(25):2419-2426. doi:10.1056/NEJMcp1113994
- Rubio-Tapia A, Hill ID, Semrad C, et al. American College of Gastroenterology Guidelines Update: Diagnosis and Management of Celiac Disease. Am J Gastroenterol. Jan 1 2023;118(1):59-76. doi:10.14309/ajg.0000000000002075
- Massironi S, Franchina M, Elvevi A, Barisani D. Beyond the gluten-free diet: Innovations in celiac disease therapeutics. World J Gastroenterol. Oct 14 2024;30(38):4194-4210. doi:10.3748/wjg.v30.i38.4194
- D’Heedene M, Vanuytsel T, Wauters L. Celiac disease: Hope for new treatments beyond a gluten-free diet. Clin Nutr. Jun 2024;43(6):1240-1249. doi:10.1016/j.clnu.2024.04.014
- Celiac Disease Foundation. Future therapies for celiac disease. Accessed August 31st, 2025. https://celiac.org/about-celiac-disease/future-therapies-for-celiac-disease/
- Vakiani E, Arguelles-Grande C, Mansukhani MM, et al. Collagenous sprue is not always associated with dismal outcomes: a clinicopathological study of 19 patients. Mod Pathol. Jan 2010;23(1):12-26. doi:10.1038/modpathol.2009.151
- Brown I, Bettington M, Rosty C. The role of histopathology in the diagnosis and management of coeliac disease and other malabsorptive conditions. Histopathology. Jan 2021;78(1):88-105. doi:10.1111/his.14262
- Weinstein WM, Saunders DR, Tytgat GN, Rubin CE. Collagenous sprue–an unrecognized type of malabsorption. N Engl J Med. Dec 10 1970;283(24):1297-301. doi:10.1056/nejm197012102832401
- Schoolmeester JK, Jenkins SM, Murray JA, Wu TT, Chandan VS. Increased immunoglobulin G4-positive plasma cells in collagenous sprue. Hum Pathol. Aug 2013;44(8):1624-9. doi:10.1016/j.humpath.2013.01.013
- Jimbo K, Aoyagi Y, Tanaka M, et al. Collagenous sprue in a 3-month-old infant. Pediatr Int. 2015;57(1):e18-22. doi:10.1111/ped.12506
- van Gils T, van de Donk T, Bouma G, van Delft F, Neefjes-Borst EA, Mulder CJ. The first cases of collagenous sprue successfully treated with thioguanine. BMJ Open Gastroenterol. 2016;3(1):e000099. doi:10.1136/bmjgast-2016-000099
- Lan N, Shen B, Yuan L, Liu X. Comparison of clinical features, treatment, and outcomes of collagenous sprue, celiac disease, and collagenous colitis. J Gastroenterol Hepatol. Jan 2017;32(1):120-127. doi:10.1111/jgh.13592
- Rubio–Tapia A, Talley NJ, Gurudu SR, Wu TT, Murray JA. Gluten-Free Diet and Steroid Treatment Are Effective Therapy for Most Patients With Collagenous Sprue. Clinical Gastroenterology and Hepatology. 2010-04-01 2010;8(4):344-349.e3. doi:10.1016/j.cgh.2009.12.023
- Jain A, Sebastian K, Quigley B. Collagenous sprue, an enigma in the spectrum of celiac disease. Clin Gastroenterol Hepatol. Jan 2014;12(1):e2-3; quiz e4-6. doi:10.1016/j.cgh.2013.05.031
- Kung VL, Liu TC, Ma C. Collagenous Enteritis is Unlikely a Form of Aggressive Celiac Disease Despite Sharing HLA-DQ2/DQ8 Genotypes. Am J Surg Pathol. Apr 2018;42(4):545-552. doi:10.1097/pas.0000000000001022
- Rubio-Tapia A, Herman ML, Ludvigsson JF, et al. Severe spruelike enteropathy associated with olmesartan. Mayo Clin Proc. Aug 2012;87(8):732-8. doi:10.1016/j.mayocp.2012.06.003
- Soendergaard C, Riis LB, Nielsen OH. Collagenous sprue: a coeliac disease look-alike with different treatment strategy. BMJ Case Rep. Mar 28 2014;2014 doi:10.1136/bcr-2014-203721
- Maguire AA, Greenson JK, Lauwers GY, et al. Collagenous sprue: a clinicopathologic study of 12 cases. Am J Surg Pathol. Oct 2009;33(10):1440-9. doi:10.1097/PAS.0b013e3181ae2545
- Zhao X, Johnson RL. Collagenous sprue: a rare, severe small-bowel malabsorptive disorder. Arch Pathol Lab Med. Jun 2011;135(6):803-9. doi:10.5858/2010-0028-rs.1
- González-Castro AM, Fernández-Bañares F, Zabana Y, et al. Microscopic Colitis and Celiac Disease: Sharing More than a Diagnostic Overlap. Nutrients. Jul 11 2024;16(14) doi:10.3390/nu16142233
- Leonard MM, Lebwohl B, Rubio-Tapia A, Biagi F. AGA Clinical Practice Update on the Evaluation and Management of Seronegative Enteropathies: Expert Review. Gastroenterology. Jan 2021;160(1):437-444. doi:10.1053/j.gastro.2020.08.061