





Cutaneous Leiomyosarcoma: Features and Management Insights
Cutaneous leiomyosarcoma: A rare cutaneous soft tissue neoplasm. Clinicopathologic Features and Review of Literature
Mukund Tinguria, MD, FRCP, FCAP1
- Department of Pathology and Laboratory Medicine, Brantford General Hospital, 200 Terrace Hill Street, Brantford, Ontario, Postal Code – N3R 1G9, Canada.
Email: [email protected]
OPEN ACCESS
PUBLISHED: 30 April 2025
CITATION: Tinguria, M., 2025. Cutaneous leiomyosarcoma: A rare cutaneous soft tissue neoplasm. Clinicopathologic Features and Review of Literature. Medical Research Archives, [online] 13(4). https://doi.org/10.18103/mra.v13i4.6437
COPYRIGHT: © 2025 European Society of Medicine. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
DOI https://doi.org/10.18103/mra.v13i4.6437
ISSN 2375-1924
ABSTRACT
Leiomyosarcomas of the skin are divided into two subtypes based on their origin and location: superficial dermal leiomyosarcoma believed to originate from the arrector pili muscles and sweat glands, and subcutaneous leiomyosarcoma arising from vascular smooth muscle of subcutaneous adipose tissue. Preoperative misdiagnosis is common because it is a rare malignant tumor, and the diagnosis is based on histopathological and immunohistochemical studies. Although superficial cutaneous leiomyosarcoma is usually treated with surgical excision, high rates of local recurrence (30–50%) have been reported. Subcutaneous leiomyosarcomas tend to be more aggressive, and since they are usually diagnosed at a more advanced stage, they are usually larger than superficial dermal based neoplasms. Recurrence rates are higher at 50–70%, and up to 60% of distant metastases have been reported. Guidelines for surgical management and role of radiation and chemotherapy as adjuvant treatments are not clearly defined. The clinicopathological features of this rare cutaneous soft tissue neoplasm are described, along with a review of the literature. Differential diagnoses, possible histogenesis, clinical behavior, management, and prognostic factors are also discussed.
Keywords: Cutaneous leiomyosarcoma, smooth muscle tumor, cutaneous neoplasm, dermatopathology.
INTRODUCTION
Cutaneous Leiomyosarcoma (cLMS) is a rare malignant neoplasm representing around 2% to 3% of all cutaneous soft tissue sarcomas and 0.04% of all neoplasms. Leiomyosarcomas of the skin can be divided into 2 subtypes based on origin and location: primary dermal leiomyosarcoma believed to originate from the arrector pili muscles and sweat glands, and subcutaneous leiomyosarcoma arising from vascular smooth muscle of subcutaneous adipose tissue. Cutaneous leiomyosarcoma and subcutaneous leiomyosarcoma differ in both primary site of origins and prognosis. Preoperative misdiagnosis is common because it is a rare malignant tumor, and direct diagnosis requires histopathologic examination. Guidelines for surgical management are not clearly defined, and recommendations for excision margins range from conservative (1 cm) to wide (2, 3, and 5 cm), including subcutaneous fat and deep fascia as well as Mohs micrographic surgery. There are no specific recommendations regarding adjuvant therapy, namely radiation and chemotherapy, for the management of cutaneous LMS.
The review highlights clinicopathologic features of cutaneous leiomyosarcoma. The review discusses the differential diagnoses, current staging system and management as well as prognostic factors. Recommendations about treatment of this rare cutaneous neoplasm is also discussed in detail.
Epidemiology
Cutaneous Leiomyosarcoma (cLMS) represents around 2% to 3% of all cutaneous soft tissue sarcomas and 0.04% of all neoplasms. On the skin, leiomyosarcoma is the third in frequency, behind dermatofibrosarcoma protuberans and Kaposi’s sarcoma. The incidence rates of cLMS have been estimated at 0.2/100,000/year.
Clinical features
Most cLMS occur in the fifth to seventh decades of life. Male to female ratio appears to be 2:3. The sites involved include head and neck, gluteal region, trunk, areola and nipple, but it frequently occurs on the lower extremities and shows a predilection toward the hair-bearing extensor surfaces. Annest et al reviewed patients with cLMS and reported that 48% of them have tumors occurring on the head and neck. The other body sites involved include scrotum, and face. Taylor et al described differences in tumor location between males and females, where females exhibited higher rates of truncal and lower extremity tumors and males had higher rates of head/ neck and upper extremity tumors.
The clinical presentation of cutaneous LMS is nonspecific. Clinically, cLMS is a slow growing lesion and appears as solitary or multiple well circumscribed nodules. Morphologic characteristics include irregular contours, pedunculation, umbilication, and reddish-brown discoloration of the skin. Cutaneous leiomyosarcoma lesions typically range from 0.3 to 3 cm in diameter. Subcutaneous lesions are usually larger, seem better delimited than dermal ones and remind a lipoma, but with more solid consistency. Cutaneous leiomyosarcomas may be asymptomatic or the patients may complain of pain, pruritus, burning sensation, and bleeding. Pain is the most common symptom of cLMS, occurring in 80% to 95% of patients. The clinical differential diagnosis includes cysts, lipomas, fibromas, persistent insect bites, granulomas, dermal nevi, pyogenic granulomas, neurofibromas, dermatofibromas, and carcinomas. In elderly patients, cutaneous leiomyosarcoma of the head and neck may clinically mimic basal cell carcinoma, squamous cell carcinoma, or pyogenic granuloma. Clinical diagnosis of cLMS is difficult. Dermoscopic features of LMS are also nonspecific, consisting of asymmetrical, ulcerated, multilobulated tumors with linear, irregular, and polymorphic atypical vessels, in addition to white structures.
Pathology
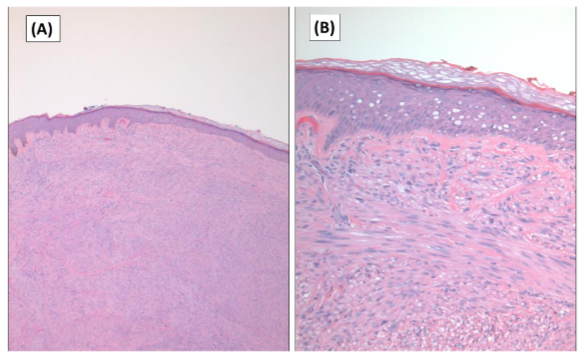
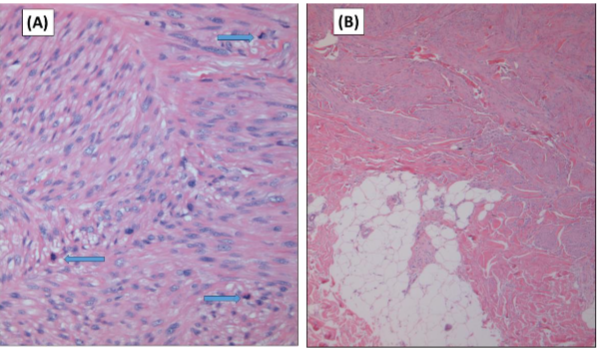
Cutaneous leiomyosarcoma is now believed to be a distinct entity separate from the subcutaneous forms, as in the past cutaneous and subcutaneous leiomyosarcomas were viewed as a single entity. Both the lesions are composed of fascicles of smooth muscle fibers. The cells are spindle-shaped, with elongated nuclei and blunt ends, inconspicuous nucleolus, and fibrillar eosinophilic cytoplasm. Some cells have a clear perinuclear halo characteristic of the muscle cell. (Figure 1). The degree of nuclear pleomorphism is variable, mitosis is usually present (>1 mitosis per 10 high-power field) and necrosis can be present or absent. The presence of cellular atypia and one mitotic figure per 10 high-power fields (HPFs) are suggestive of LMS according to the WHO recommendations. Ki-67 can be used to assess proliferative activity in difficult cases. Phosphohistone H3 (PHH3) immunostains may also be employed to identify mitotic figures. Two architectural patterns have been described: the nodular pattern, characterized by greater cellularity, atypia and mitotic figures, and the diffuse pattern, which is less cellular and with a lower mitotic burden. Occasionally, there is no obvious cellular atypia, making a differential diagnosis with leiomyoma difficult. In these instances, the histological diagnosis should be based on the overall architecture of the lesion, with an infiltrative pattern and increased cellularity being suggestive of malignancy. Rare histologic variants of leiomyosarcoma include clear cell variant, desmoplastic leiomyosarcoma, LMS of epithelioid cells, LMS of multinucleated giant cells, LMS of granular cells, sclerotic LMS, and pleomorphic and myxoid variants.
Differential Diagnosis
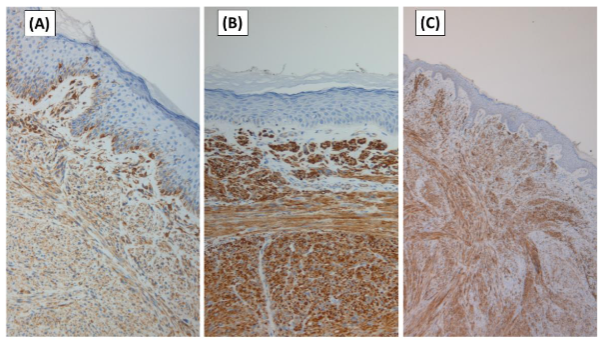
Entities, which may have overlapping features with leiomyosarcoma include spindle cell squamous cell carcinoma, atypical fibroxanthoma (AFX), pleomorphic undifferentiated sarcoma, desmoplastic malignant melanoma, cellular neurofibroma, benign fibrous histiocytoma/dermatofibroma, cellular dermatofibroma, dermatofibrosarcoma protuberans and leiomyoma. Immunohistochemical analysis is essential for accurately diagnosing muscle-specific antigens and facilitating differentiation of smooth muscle tumors from malignant spindle cell cutaneous tumors. Cutaneous leiomyosarcomas are generally positive for vimentin, desmin, smooth muscle actin, HHF. Vimentin and smooth muscle actin positivity is noted in 100% of cLMS, and desmin, in 60% of cLMS. Actin expression may be a more sensitive marker of myogenic differentiation than desmin. Absence of desmin expression has been observed in poorly differentiated neoplasms and may represent a feature of neoplasms that arise from vessel walls. Cytokeratin and S100 stains may rarely be positive, causing difficulty in differentiating these tumors from other spindle cell neoplasms such as squamous cell carcinoma and malignant melanoma. The positivity of p53 has been demonstrated in some studies. Diffuse overexpression of p53 has been described in cutaneous LMS but not in leiomyomas, being helpful in the differential diagnosis between these two conditions. The loss of PTEN was noted by Hall et al, but its significance is unknown. Estrogen and progesterone receptors were also expressed by some soft tissue LMS. Other immunohistochemical stains performed to rule out other spindle-cell lesions (spindle cell carcinoma, desmoplastic melanoma, dermatofibrosarcoma protuberans, nodular fasciitis, malignant peripheral nerve sheath tumor, spindle cell atypical fibroxanthoma, fibrosarcoma, synovial sarcoma, and vascular tumors) include EMA, CD34, CD117, CEA, HMB45, Mart-1, Melan A and CK7; all of them are negative in LMS.
Staging
There are currently two staging systems for LMS. The Federation Nationale des Centres de Lutte Contre Le Cancer (FNCLCC) grading system is determined by three parameters, including differentiation, mitotic activity, and extent of necrosis (Table 1) and the American Joint Committee on Cancer (AJCC) Cancer Staging. Recent literature demonstrated the utility of the American Joint Committee on Cancer GTNM (Grade, Tumor, Nodes, Metastasis) staging criteria for soft tissue sarcoma in the assessment of both forms of superficial leiomyosarcoma. Recently, multivariate analysis of 105 cases of superficial leiomyosarcoma (both cutaneous and subcutaneous types) demonstrated that AJCC stage and overall tumor size were the only statistically significant prognosticators of 5-year patient survival.
| Tumor Differentiation | Grade |
|---|---|
| Sarcoma closely resembling normal adult mesenchymal tissue | 1 |
| Sarcoma for which histologic typing is certain | 2 |
| Embryonal and undifferentiated sarcoma, sarcoma of uncertain type, synovial sarcoma, soft tissue osteosarcoma, and Ewing sarcoma/primitive neuroectodermal tumor (PNET) of soft tissue | 3 |
| T – Tumor | Classification |
|---|---|
| Tx | Primary tumor cannot be assessed |
| T0 | No evidence of primary tumor |
| T1 | Tumor ≤5 cm in greatest dimension |
| T2 | Tumor >5 cm and ≤10 cm in greatest dimension |
| T3 | Tumor >10 cm and ≤15 cm in greatest dimension |
| T4 | Tumor >15 cm in greatest dimension |
| Stage | Classification |
|---|---|
| IA | T1 N0 M0 G1, GX |
| IB | T2, T3, T4 N0 M0 G1, GX |
| II | T1 N0 M0 G2, G3 |
| IIIA | T2 N0 M0 G2, G3 |
| IIIB | T3, T4 N0 M0 G2, G3 |
| IV | Any T N1 M0 Any G |
| IV | Any T Any N M1 Any G |
Treatment / Management
Complete excision with clear margins is considered the gold standard. Recommendations or guidelines for the size of margin are, however, lacking. Margins of 3–5 cm have been proposed by some investigators for both cutaneous LMS and subcutaneous LMS. In the study by Deneve et al., excision, with a 1 cm margin was performed for 33 cases of cutaneous LMS, with no recurrence observed in more than 15 months of follow-up. It is important to note that, however, the average size of lesions included was 1.3 cm, and 88% were low-grade histologically. There are no reports on dependence of either the survival benefit or the rate of local recurrence on the excision width. The rate of recurrence has been claimed to decrease with wider excision, but there are no documented figures for these claims. If microscopic margins are negative for soft tissue sarcomas, then local recurrence and survival are not affected by increasing the soft tissue excisional margin. The deep margin should reach up to the fascia, and in more infiltrating cases the muscle should be included. It is estimated that up to 20% of patients may require repeated excisions to achieve negative margins. MMS, a time-honored tissue-conserving technique that allows for optimal cosmetic outcome, has been shown to be an equally effective, if not superior, modality in treating spindle cell neoplasms, including cutaneous LMS. Kazlouskaya et al. reviewed 47 cases of cutaneous LMS treated with MMS. There were only two cases of recurrences and one case of metastases identified among 47 cases of LMS treated with MMS. MMS has a distinct advantage over surgical excision in certain cases, i.e., a simultaneous closure can be performed after confirming clear margins. Despite this, MMS is still underutilized for cutaneous LMS, with only about 5% of cases being treated with this method.
There are no specific recommendations regarding adjuvant therapy, namely radiation and chemotherapy, for the management of cutaneous LMS. Indications for adjuvant chemoradiotherapy include tumor size greater than 5 cm, positive excision margins, high grade of atypia, local recurrence, as well as the presence of metastasis. Radiotherapy can also be used in local palliative control in cases with metastasis. No clear survival benefit has been demonstrated with the use of adjuvant chemotherapy or radiotherapy; however, external beam radiation is commonly administered in cases of high-grade tumors. Advanced therapies, such as immunotherapy and gene therapy, are being developed. Targeted therapies using tyrosine kinase inhibitors are also currently being investigated in clinical trials.
Clinical Behaviour and Prognosis
The reported biologic behavior of superficial leiomyosarcoma varies widely in the literature. This is due to three factors. First, these tumors are extremely rare. Second, calculations and reports include cutaneous lesions only in some cases, subcutaneous lesions only in others, and an admixture of both lesions in yet other reports. Many published series do not specify surgical margins, listing only “excised” or “wide excision” as tumor treatments, henceforth creating difficulty in systematically reviewing treatment responses and overall biologic behavior. Jensen et al described several poor prognostic factors, including tumor size more than 5 cm, deep location with fascia involvement, high malignancy grade, and acral distribution, and their only patient with local recurrence as having incomplete microscopic margins at initial surgery.
The clinical behavior is correlated to the depth of invasion with cLMS demonstrating a more benign tumor biology than leiomyosarcoma (LMS) arising within the subcutaneous tissue. It is therefore critical to evaluate the entire depth of the tumor to assess its origin. Based on the low rate of recurrence and absence of metastases, the name “atypical intradermal smooth muscle neoplasm” was proposed in 2011 for these tumors. The local recurrence rate for superficial cutaneous leiomyosarcoma is approximately 30%; however, reported rates range widely from 0% to 50% depending upon treatment modality and duration of follow-up. By contrast, subcutaneous leiomyosarcomas have a much higher rate of local recurrence. These tumors recur locally in 50% to 70% of cases. Inability to achieve clear margins is one of the most important factors in predicting recurrence. Oliver et al proposed that the risk of local recurrence is related to the adequacy of excision, whereas metastatic spread is related to depth, histologic grade of the tumor, and DNA content.
The metastatic potential of cutaneous and subcutaneous leiomyosarcoma is also dramatically different. Cutaneous LMS rarely metastasizes and has a favorable prognosis overall. Kraft and Fletcher reported no metastases among 83 cases of cutaneous LMS and proposed that the term “sarcoma” be omitted altogether. Other studies reported a rate of metastasis of up to 12% for cutaneous LMS. The metastatic rate for subcutaneous LMS, in contrast, is much higher, ranging from 27.3% to 63%. Higher histological grade, size greater than 5 cm, presence of necrosis, and deeper location increase the likelihood of cutaneous LMS metastases. Distant metastases are most commonly observed in the lungs, but superficial LMS may also metastasize to other cutaneous sites, namely to the scalp; scalp metastases may even be a harbinger of poor outcomes.
Only few cases of mortality have been associated with cutaneous LMS. Fauth et al. reported only one patient with cutaneous LMS (1/14; 7.14%) developing lung metastases and subsequently expiring from the disease in about a year. Similarly, Winchester et al. reported two deaths out of 48 cases with cutaneous LMS (4.2%). On the contrary, subcutaneous LMS has a significantly higher mortality rate that ranges from 25% to 41.2%. The Scandinavian Sarcoma Research Group has estimated the overall survival rate for LMS to be 64% for 5 years and 46% for 10 years, with the presence of metastases being the most important prognostic factor. Bresler et al in their retrospective analysis of 36 cases of subcutaneous LMS reported metastasis in 12 cases (33%) with lung being the most common site of metastasis. Six of 36 patients (17%) died from the disease at an average of 47 months after diagnosis (range 16 to 94 months). Metastasis or death from disease was significantly associated with the Fédération Nationale des Centres de Lutte Contre le Cancer grade (P = 0.0015), the presence of necrosis (P = 0.032), tumor size (P = 0.049), and AJCC tumor stage (P = 0.036). These data demonstrate that subcutaneous LMS are more aggressive than dermal-based tumors and have a prognosis akin to that of deep-seated LMS. The prognosis of dermal LMS is usually better, with 5-year-survival rates over 95%. Subcutaneous LMS has a greater tendency for recurrence and metastasis, therefore resulting in lower 5-year-survival rates of around 65%. Wong et al. retrospectively reviewed all patients with dermal and subcutaneous leiomyosarcoma managed at the Peter MacCallum Cancer Centre, Australia from January 2003 to December 2018. Eighty-three patients were identified (64 with dermal leiomyosarcomas and 19 with subcutaneous leiomyosarcomas). Dermal leiomyosarcoma had an excellent prognosis, particularly after definitive surgical excision, with margins of at least 10 mm. Subcutaneous leiomyosarcoma had poorer outcomes and should be managed by wider excision and considered for adjuvant radiotherapy. In a multivariate analysis of prognostic factors for superficial LMS, such as size, depth of invasion, histological grade and proliferative activity (Ki67), only tumor size was shown to be an independent prognostic factor in relation to decreased survival. In this study and in a univariate analysis, a tumor size equal to or greater than 5 cm, deep location with fascia involvement and a high histological grade have been correlated with decreased survival. Additionally, in subsequent studies, histological grades II and III (according to the Fédération Nationale des Centres de Lutte le Cancer, or FNCLCC grading system) have been associated with recurrence and metastasis.
Fernandez reviewed the literature to study the evolution and prognostic factors of primary and cutaneous leiomyosarcoma. The published data were reviewed to assess the rates of recurrence and metastasis, mortality, recommended follow-up periods, and prognostic factors. This review included 112 subcutaneous LMS and 313 cutaneous LMS cases. In subcutaneous LMS, the rates of recurrence, metastasis, and mortality were 36.63%, 43.23%, and 37.82%, respectively, after a median follow-up period of 4.40 years, while in cutaneous LMS, these rates were 24.40%, 4.22%, and 3.33%, respectively, after a median follow-up period of 3.45 years. The cutaneous LMS have a potential risk of metastasis, and in certain cases, they may be the cause of death. The collected data suggest that the term “atypical intradermal smooth neoplasm” should be avoided to identify dermal LMS.
Taylor et al. explored the presentation of LMS and disease-specific survival (DSS) with respect to sex. Males demonstrated higher 5- and 10-year DSS rates (69.0% and 60.0%, respectively) than females (50.0% and 36.0%) (p < 0.001). Multivariate analysis adjusted for age, race, ethnicity, income, rural–urban living, disease stage, and primary tumor location revealed that female sex independently increased disease-specific mortality risk compared to male sex.
There are no standard guidelines for LMS follow-up, but clinical examination is recommended every 4 months during the first two years, for early detection of possible local recurrences. Thereafter, controls every 6 months until the fifth year after surgery are advised; subsequently, once a year until 20 years, since very late relapses have been described. However, the practice of a simple annual chest radiograph in the first five years after surgery and clinical evaluation of the surgical bed and regional lymph nodes appears to be a reasonable strategy. In some cases, magnetic resonance imaging may be helpful, especially in recurrent or hypodermic lesions, or in cases where surgery is complex.
Histogenesis / Genetics
Most cLMS arise de novo rather than via malignant transformation of leiomyoma as a precursor lesion. Although the etiology of cutaneous leiomyosarcoma is unclear, predisposing factors such as exposure to trauma, radiation, chemicals, and sunlight have been emphasized. Recently, cLMS arising from a tattoo, pacemaker pocket, or scar has been reported. In immunocompromised pediatric patients, including those with HIV, an increased incidence of LMS was associated with Epstein-Barr virus infection.
Although the underlying genetic abnormalities responsible for the development of cutaneous LMS are poorly understood, one study has identified defects in the 13q4-q21 chromosome region. LMS can also be seen in the setting of Reed syndrome, with fumarate hydratase gene mutation being implicated as an important factor. Moreover, aberrations in the p53 and p16 genes may play a role and have been shown to be associated with poor prognosis. Kraft and Fletcher described a case of LMS and pleomorphic liposarcoma developing in a patient with Li–Fraumeni syndrome, a hereditary disorder associated with a germline mutation in the p53 tumor suppressor gene.
A 32-year-old patient with Li–Fraumeni syndrome, who developed cutaneous LMS with a hereditary disorder associated with a germline mutation in the p53 tumor suppressor gene.
Conclusion
Leiomyosarcoma of the skin is a rare tumor of smooth muscle derivation that is diagnosed by histopathological and immunohistochemical studies. There are two subtypes of superficial leiomyosarcoma: cutaneous and subcutaneous. Although cutaneous LMS has a better prognosis than subcutaneous LMS, it is unclear whether larger tumors or those that secondarily involve the subcutis exhibit different behaviors. The AJCC staging criteria for soft tissue sarcomas provide valuable prognostic information for cutaneous leiomyosarcomas. Although surgical excision is regarded as the mainstay treatment for cutaneous LMS, more prospective studies are needed to standardize the margins. The clear differences in biological behavior between cutaneous and subcutaneous tumors provide compelling evidence that not all types of superficial leiomyosarcomas should be treated equally, with more aggressive treatment warranted only in subcutaneous tumors. Although MMS can be a promising surgical modality, fewer than 50 cases have been performed, with insufficient follow-up to demonstrate its long-term effectiveness. MMS may be safely introduced for small cutaneous LMS; however, the tumor characteristics and patient selection criteria remain to be established. Although radiation and chemotherapy may be employed as adjuvant treatments for larger, recurrent, and subcutaneous LMS, the optimal regimens still need to be elucidated. Adjuvant therapy may be employed, after multidisciplinary discussions until specific guidelines for adjuvant therapy are established. There are no standard guidelines for LMS follow-up, and further studies are required to establish follow-up guidelines.
Conflict of Interest:
None.
Funding Statement:
None.
Acknowledgements:
None.
References:
- Rodríguez-Lomba E, Molina-López I, Parra-Blanco V. et al. Clinical and histopathologic findings of cutaneous leiomyosarcoma: correlation with prognosis in 12 patients. Actas Dermosifiliogr. 2018;109:140 – 7.
- Stout A, Hill W. Leiomyosarcoma of the superficial soft tissue. Cancer 1964; 11: 844.
- Liao W, Wang Y, Ma H. Cutaneous leiomyosarcoma: the clinical experience of Taipei veterans general hospital revisited. Ann Plast Surg 2017; 78(3 Suppl 2): S47–S51.
- Massi D, Franchi A, Alos L, et al. Primary cutaneous leiomyosarcoma: clinicopathological analysis of 36 cases. Histopathology. 2010;56:251–262.
- Kraft S, Fletcher C. Atypical intradermal smooth muscle neoplasms: clinicopathologic analysis of 84 cases and a reappraisal of cutaneous “leiomyosarcoma”. Am J Surg Pathol. 2011;35:599–607.
- Fields J, Helwig E. Leiomyosarcoma of the skin and subcutaneous tissue. Cancer. 1981;47:156–169.
- Snowden R, Osborn F, Wong F. et al. Superficial leiomyosarcoma of the head and neck: case report and review of the literature. Ear Nose Throat J. 2001; 80; 449–453.
- Kaddu S, Beham A, Cerroni L. et al. Cutaneous leiomyosarcoma. Am. J. Surg. Pathol. 1997; 21; 979–987.
- Annest N, Grekin S, Stone M. et al. Cutaneous leiomyosarcoma: a tumor of the head and neck. Dermatol. Surg. 2007; 33; 628–633.
- González-Sixto B, De la Torre C, Pardavila R. et al. Leiomyosarcoma arising from scrofuloderma scar. Clin Exp Dermatol. 2008;33:776–778.
- West C, Morritt A, Pedelty L. et al. Cutaneous leiomyosarcoma arising in a tattoo—‘a tumour with no humour’. J Plast Reconstr Aesthet Surg. 2009;62: e79.
- González-Vela M, Val-Bernal J, Rubio S. et al. Cutaneous leiomyosarcoma developing on a pacemaker pocket. Dermatol Surg. 2009;35:863–867.
- Pol R, Dannenberg H, Robertus J. et al. Cutaneous leiomyosarcoma arising in a smallpox scar. World J Surg Oncol. 2012;10:148.
- Weiss S, Goldblum J. Enzinger and Weiss’s Soft Tissue Tumors. 6th ed. Saunders; 2013.
- Holst V, Junkins-Hopkins J, Elenitsas R. Cutaneous smooth muscle neoplasms: clinical features, histologic findings, and treatment options. J Am Acad Dermatol. 2002;46:477–490.
- Pijpe J, BroersG, Plaat B. et al. The relation between histological, tumor biological and clinical parameters in deep and superficial leiomyosarcoma and leiomyoma. Sarcoma. 2002;6:105–110.
- Fons M, Bachhuber T, Plaza J. Cutaneous leiomyosarcoma originating in a symplastic pilar leiomyoma: a rare occurrence and potential diagnostic pitfall. J Cutan Pathol. 2011;38:49–53.
- Winchester D, Hocker T, Brewer J. et al. Leiomyosarcoma of the skin: clinical, histopathologic, and prognostic factors that influence outcomes. J Am Acad Dermatol. 2014;71:919–925.
- Blaise G, Nikkels A. Childhood cutaneous leiomyosarcoma. Pediatr Dermatol. 2009; 26:477–479.
- Bellezza G, Sidoni A, Cavaliere A. et al. Primary cutaneous leiomyosarcoma: a clinicopathological and immunohistochemical study of 7 cases. Int J Surg Pathol. 2004;12:39–44.
- Bernstein S, Roenigk R. Leiomyosarcoma of the skin. Treatment of 34 cases. Dermatol Surg. 1996; 22:631–635.
- Yanguas I, Goday J, González-Güemes M. et al. Cutaneous leiomyosarcoma in a child. Pediatr Dermatol. 1997; 14:281–283.
- Miyajima K, Oda Y, Oshiro Y. et al. Clinicopathological prognostic factors in soft tissue leiomyosarcoma: a multivariate analysis. Histopathology. 2002;40: 353–359.
- Oliver G, Reiman H, Gonchoroff N. et al. Cutaneous and subcutaneous leiomyosarcoma: a clinicopathological review of 14 cases with reference to antidesmin staining and nuclear DNA patterns studied by flow cytometry. Br J Dermatol. 1991;124:252–257.
- Christopher J, Porter M, Januszkiewicz J. Cutaneous leiomyosarcoma. Plast Reconstr Surg. 2002;109:964–967.
- Iacobucci J, Stevenson T, Swanson N, et al. Cutaneous leiomyosarcoma. Ann Plast Surg. 1987;19:552–554.
- Lin J, Tsai R. Subcutaneous leiomyosarcoma on the face. Dermatol Surg. 1999;25:489–491.
- Tsutsumida A, Yoshida T, Yamamoto Y. Management of superficial leiomyosarcoma: a retrospective study of 10 cases. Plast Reconstr Surg. 2005; 116:8–12.
- Starling J, Coldiron BM. Mohs micrographic surgery for the treatment of cutaneous leiomyosarcoma. J Am Acad Dermatol. 2011;64:1119–1122.
- Deneve J, Messina J, Bui M, et al. Cutaneous leiomyosarcoma: treatment and outcomes with a standardized margin of resection. Cancer Control. 2013; 20:307–312.
- Llombart B, Serra-Guillén C, Requena C. et al. Leiomyosarcoma and pleomorphic dermal sarcoma: guidelines for diagnosis and treatment. Actas Dermosifiliogr. 2019;110:4 – 11.
- Kohlmeyer J, Steimle-Grauer S, Hein R. Cutaneous sarcomas. J Dtsch Dermatol Ges. 2017;15:630 – 48.
- John T, Portenier D, Auster B. et al. Leiomyosarcoma of the scrotum – a case report and review of the literature. Urology 2006;67(2):424e13–15.
- Torres T, Oliveira A, Sanches M. et al. Superficial cutaneous leiomyosarcoma of the face: report of three cases. J Dermatol. 2011;38:373– 376.
- Taylor M, Thomas S, Ituarte B. et al. Sex disparities in leiomyosarcoma of the skin: females experience worse disease-specific survival. Archives of Dermatological Research (2024) 316:469.
- Zaballos P, Del Pozo L, Argenziano G. et al. Dermoscopy of cutaneous smooth muscle neoplasms: a morphological study of 136 cases. J Eur Acad Dermatol Venereol 2019; 33: 693–699.
- Dahl I, Angervall L. Cutaneous and subcutaneous leiomyosarcoma: a clinicopathologic study of 47 patients. Pathol Eur. 1974;9: 307–315.
- Davidson L, Frost M, Hanke W. et al. Primary leiomyosarcoma of the skin: case report and review of the literature. J Am Acad Dermatol 1989;21:1156–60.
- Aneiros-Fernandez J, Antonio Retamero J, Husein-Elahmed H. et al. Primary cutaneous and subcutaneous leiomyosarcomas: review of their evolution and prognostic factors. Eur J Dermatol 2016; 26(1): 9-12 doi:10.1684/ejd.2015.2681.
- Weedon D, Williamson R, Patterson J. Smooth and skeletal muscle tumours. World Health Organization classification of tumours. Pathology & genetics. Skin tumours. Lyon: IARC Press, 2006; 250–253.
- Idriss M, Kazlouskaya V, Malhotra S. et al. Phosphohistone-H3 and Ki-67 immunostaining in cutaneous pilar leiomyoma and leiomyosarcoma (atypical intradermal smooth muscle neoplasm). J Cutan Pathol 2013; 40: 557–563.
- Atzori L, Pilloni L, Zanniello R. et al. Clear-cell variant of superficial cutaneous leiomyosarcoma associated with RB1 mutation: Clinical, dermoscopic, and histopathological characteristics. J Cutan Pathol. 2020;47:571–575.
- Choy C, Cooper A, Kossard S. Primary cutaneous diffuse leiomyosarcoma with desmoplasia. Australas J Dermatol 2006; 47: 291–295.
- Karroum J, Zappi E, Cockerell C. Sclerotic primary cutaneous leiomyosarcoma. Am J Dermatopathol 1995; 17: 292–296.
- Diaz-Cascajo C, Borghi S, Weyers W. Desmoplastic leiomyosarcoma of the skin. Am J Dermatopathol 2000; 22:251–255.
- Berzal-Cantalejo F, Sabater-Marco V, Perez-Valles A. et al. Desmoplastic cutaneous leiomyosarcoma: case report and review of the literature. J Cutan Pathol 2006; 33(Suppl 2): 29–31.
- Fernandez-Flores A. Cutaneous leiomyomas and leiomyosarcomas: an immunohistochemical study with p53. Rom J Morphol Embryol 2010; 51: 295–298.
- Hall B, Grossmann A, Webber N. et al. Atypical intradermal smooth muscle neoplasms (formerly cutaneous leiomyosarcomas): case series, immunohistochemical profile and review of the literature. Appl Immunohistochem Mol Morphol 2013; 21: 132–138.
- Carvalho J, Thomas D, Lucas D. Cluster analysis of immunohistochemical markers in leiomyosarcoma delineates specific anatomic and gender subgroups. Cancer 2009; 115: 4186–4195.
- Trojani M, Contesso G, Coindre J, et al. Soft-tissue sarcomas of adults; study of pathological prognostic variables and definition of a histopathological grading system. Int J Cancer 1984; 33: 37–42.
- Amin M, Edge S, Greene F. et al. AJCC Cancer Staging Manual, 8th edn. Cham, Switzerland: Springer International Publishing, 2017.
- Jensen M, Jensen O, Michalski W. et al. Intradermal and subcutaneous leiomyosarcoma: a clinicopathological and immunohistochemical study of 41 cases. J Cutan Pathol. 1996; 23:458–463.
- Glazer E, Prieto-Granada C, Zager J. Current approaches to cutaneous sarcomas: dermatofibrosarcoma protuberans and cutaneous leiomyosarcoma. Curr Probl Cancer 2015; 39: 248– 257.
- Feigenbaum L, Skinner D, Golda N. Atypical cutaneous leiomyosarcoma with skip-lesion behavior. Dermatol Surg 2013;39: 660–662.
- Vujevich J, Goldberg L, Kimyai-Asadi A. et al. Recurrent nodule on the nasal columella: a good reason to re-biopsy. Int J Dermatol 2008; 47: 728–731.
- Fauth C, Bruecks A, Temple W. et al. Superficial leiomyosarcoma: a clinicopathologic review and update. J Cutan Pathol 2010; 37: 269–276.
- Huether M, Zitelli J, Brodland D. Mohs micrographic surgery for the treatment of spindle cell tumors of the skin. J Am Acad Dermatol 2001; 44: 656–659.
- Wollina U, Koch A, Hansel G. et al. A 10-year analysis of cutaneous mesenchymal tumors (sarcomas and related entities) in a skin cancer center. Int J Dermatol 2013; 52: 1189–1197.
- Humphreys T, Finkelstein D, Lee J. Superficial leiomyosarcoma treated with Mohs micrographic surgery. Dermatolo Surg 2004; 30: 108–112.
- Kazlouskaya V, Lai Y, Khachemoune A. Leiomyosarcoma of the skin: review of the literature with an emphasis on prognosis and management. International Journal of Dermatology. Feb. 2020;59(2):165-172.
- Ghareeb E, Dulmage B, Vargo J. et al. Underutilization of Mohs micrographic surgery for less common cutaneous malignancies in the United States. Dermatol Surg 2016; 42: 653–662.
- Svarvar C, Böhling T, Berlin Ö. et al. Clinical course of nonvisceral soft tissue leiomyosarcoma in 225 patients from the Scandinavian sarcoma group. Cancer. 2007;109:282–291.
- Wascher R, Lee M. Recurrent cutaneous leiomyosarcoma. Cancer 1992; 70; 490–492.