





DNA Methyltransferase Inhibitors in Triple Negative Breast Cancer
The promise of DNA methyltransferase inhibitors for the treatment of triple negative breast cancer
Daifuku R.
- Epigenetics Pharma. 244 Davison Head Dr. Friday Harbor, WA 98250. USA. Email: [email protected]
OPEN ACCESS
PUBLISHED: 28 February 2025
CITATION: Daifuku, R., 2025. The promise of DNA methyltransferase inhibitors for the treatment of triple negative breast cancer. Medical Research Archives, [online] 13(2). https://doi.org/10.18103/mra.v13i2.6276
COPYRIGHT: © 2025 European Society of Medicine. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
DOI https://doi.org/10.18103/mra.v13i2.6276
ISSN 2375-1924
Abstract
In cancer, loss of gene expression occurs about 10 times more frequently because of hypermethylation than mutations. Hypermethylation can inhibit genes that prevent cancer formation and genes responsible for an antitumor immune response, and is particularly common in triple negative breast cancer (TNBC). The reversibility of these covalent modifications makes them attractive targets for therapeutic intervention. There are two approved DNA methyltransferase (DNMT) inhibitors for the treatment of hematologic malignancies, 5-azacytidine and decitabine. Unfortunately, these two drugs have not yet demonstrated efficacy in the treatment of breast cancer (BC). iMN013 is a nucleoside that has a base identical to decitabine and 5-azacytidine that confers DNMT inhibition, leading to the expression of hypermethylated genes. The sugar is identical to that of gemcitabine, a drug approved for the treatment of metastatic BC that inhibits ribonucleotide reductase, a critical target for TNBC survival. iMN013 is active against BC cell lines in the nM range. The prodrug of iMN013, iMN041 was demonstrated to be an immunotherapeutic in a syngeneic tumor mouse model with significant increases in granzyme B in NK and NKT cells, and in the ratios of CD8-T to regulatory T cells (Treg) and CD4-T to Treg cells while inhibiting myeloid-derived suppressor cells, compared to vehicle controls. Decreased numbers of Treg cells were noted in iMN041 treated animals, differing from decitabine which has been shown to stimulate an increase in Treg cells. A significant decrease in MAGE-A positive tumor cells in iMN041 treated mice suggests that these cells are one of the main targets of the activated immune system. Mage-A is frequently elevated in TNBC. iMN041 was effective in mouse xenograft models of solid tumors, including non-small cell lung, pancreatic, renal and TNBC. In the TNBC model (cell line DU4475), iMN041 demonstrated significant inhibition of tumor growth and improved survival of treated mice compared to vehicle control. iMN041 raises the possibility of treating TNBC patients with a more effective and less toxic regimen that will favorably impact survival by derepressing genes, such as the tumor suppressor TP53, resulting in tumor cell apoptosis, and by stimulating of the own anti-tumor immune response.
Keywords
DNA methyltransferase inhibitors, triple negative breast cancer, immunotherapy, iMN013, iMN041
Introduction
About 1 in 8 U.S. women will develop invasive breast cancer (BC) over the course of her lifetime. An estimated 272,454 new cases of BC in women were diagnosed in 2021 in the U.S. and 42,211 women died in 2022. BC is categorized into three major subtypes based on the presence or absence of molecular markers for estrogen (ER) or progesterone receptors (PR) and human epidermal growth factor 2 (ERBB2): hormone receptor (+)/ERBB2 (-), ERBB2 (+), and triple-negative tumors (TNBC) lacking all 3 standard molecular markers (15% of patients). Most new treatment options for metastatic breast cancer (mBC) recently approved by the Food and Drug Administration (FDA) are only effective for ER/PR (+) or ERBB2 (+) metastatic tumors, and the subset of patients with metastatic TNBC (mTNBC) is not sensitive to endocrine therapy or molecular targeted therapy. Chemotherapy remains the main systemic treatment, but the efficacy of conventional postoperative adjuvant chemoradiotherapy is poor. There has been some progress in tailoring drugs and regimens to specific patients with mTNBC, but only 30% of patients can achieve a complete response. Median overall survival for mTNBC is approximately 1 year vs. approximately 5 years for the other 2 subtypes.
Various therapeutic approaches are under development, such as therapies targeting DNA repair pathways, androgen receptor signaling pathways, and kinases. In addition, immunotherapy has also been demonstrated to improve overall survival and response in TNBC. Another promising approach under evaluation is the treatment of TNBC patients with a DNA methyltransferase inhibitor (DNMTI). A review of current research on treatment of TNBC with DNMTIs is presented herein, as is the therapeutic promise of a novel DNMTI for the treatment of solid tumors, iMN013, and its prodrug iMN041.
Background on DNA methyltransferase inhibitors
There is increasing interest in the possibility of treating mBC with a DNMTI, particularly mTNBC. DNA methylation is highly dysregulated in cancer with hypomethylation of distal regulatory regions and repetitive elements, and hypermethylation of CpG (cytidine-phosphate-guanine dinucleotide) islands in promoter regions. DNA methyltransferases (DNMTs) are a family of enzymes, the most prevalent of which is DNMT1, with DNMT3a and DNMT3b being barely detectable. DNMT1 and DNMT3b overexpression can lead to inactivation of gene expression that suppresses tumorigenesis. These genes include (1) tumor suppressor genes such as TP53, (2) genes that promote apoptosis, and suppress metastasis and angiogenesis, (3) DNA repair genes, and (4) genes that express tumor-associated antigens. Since this epigenetic change is reversible, it presents an attractive target for therapeutic intervention.
The most effective DNMTIs remain those nucleosides having a 5-azacytosine base, and include the two approved DNMTIs, decitabine and 5-azacytadine (azaC), as well as a recently developed nucleoside, iMN013, and its prodrug iMN041. Decitabine has been shown to inhibit DNMT1, DNMT3a and DNMT3b. Both azaC and decitabine are DNMTIs approved in the U.S. for the treatment of myelodysplastic syndrome (MDS). More recently, decitabine has been approved in the European Union for the treatment of acute myeloid leukemia (AML). The benefit of current DNMTIs in solid tumors is not as well established and in a meta-analysis of clinical trials in such patients, DNMTIs were shown to be able to improve clinical outcome, although overall response was limited. Indeed, clinical trials exploring DNMTI monotherapy in BC have not shown significant benefits to date.
DNA methyltransferase inhibitors have demonstrated the ability to derepress important genes in breast cancer
TNBC promoter hypermethylation events are more frequent compared with other BC subtypes, suggesting that targeting DNMT may have clinical benefits for TNBC patients. Decitabine has been shown to induce degradation of DNMT1, DNMT3a, and DNMT3b by TRAF6 through a lysosome-dependent protein-degradation pathway and inhibits tumor growth, particularly in tumors with high DNMT protein levels. In a TNBC patient derived xenograft (PDX) model, tumors responded to decitabine regardless of chemotherapy response. Thus, TNBC patients with high DNMT levels and resistance to standard chemotherapy may still benefit from treatment with a DNMTI.
DNA hypermethylation, primarily mediated by aberrant expression of DNMT1, has been associated with poor survival of TNBC patients. The oncogenic effects of DNMT1 in TNBC include: (1) inhibition of ER expression, (2) promotion of epithelial-mesenchymal transition required for metastasis, (3) induction of cellular autophagy and (4) promotion of the growth of cancer stem cells. Data suggesting that ER is silenced epigenetically has led to attempts to re-express ER and restore its functionality and hormonal sensitivity to provide a therapeutic target for ER-negative BC. One study demonstrated that treatment of ER-α negative BC cells with decitabine, led to the re-expression of ER mRNA and functional ER proteins by specifically inhibiting DNMT1 expression.
The expression of DNMT1 or DNMT3a was significantly correlated with metastasis in patients with BC, and the expression of DNMT3a and 3b was significantly correlated with advanced clinical stages. Overexpression of DNMT1 or DNMT3a led to promoter hypermethylation and reduced expression of BRCA1. BRCA1 is one of the most common tumor suppressor genes, and plays an important role in DNA repair, replication fork protection, cell cycle regulation, and gene transcription regulation. DNMT3a-mediated hypermethylation can suppress several critical genes in BC, including SOX2 and HIF-1α. SOX2 plays a critical role in the migration, proliferation and invasion of BC cells, while HIF-1α is the regulator of cellular adaptation to hypoxia in rapidly growing tumors.
PKD1 is a serine-threonine kinase expressed in ductal epithelial cells and a negative regulator of actin reorganization processes necessary for cell migration and invasion. Expression is lost during BC progression to an aggressive metastatic phenotype, and mediated by hypermethylation and inactivation of its promoter. In vitro, treatment with a DNMTI has been shown to derepress PKD1. Lysophosphatidic acid receptor 6 (LPAR6) expression is significantly reduced in BC. Decitabine was found to upregulate LPAR6, a possible tumor suppressor, in 98 samples of clinical BC. LRIG1 (leucine-rich repeat and immunoglobulin-like domain) is a negative regulator of receptor tyrosine kinases and a tumor suppressor. Across BC subtypes, LRIG1 expression is lowest in TNBC subtypes. LRIG1 CpG island methylation is most prominent in TNBC lines and patient samples. Decitabine leads to increased LRIG1 transcript expression in TNBC cell lines, while having no effect on LRIG1 expression in luminal/ER-positive cell lines.
DNA methyltransferase inhibitors play a role in sensitizing breast cancer cells to other drugs
Numerous examples highlight the association between resistance to specific therapies and changes in DNA methylation patterns. For example, trastuzumab resistance correlates with hypermethylation of the TGFB1 gene. Similarly, resistance to doxorubicin has been linked with hypermethylation of the MSH2 gene in BC cells. In a study of 23-genes, a chemoresistance signature was developed from chemoresistant BC and was associated with poor prognosis with multiple chemotherapeutic agents. Of several compounds, decitabine was found to be the compound most likely to target the signature, and in BC cell lines the signature-related chemoresistance could be suppressed by decitabine treatment. The cytotoxicity of paclitaxel, adriamycin, and 5-fluorouracil was analyzed against human BC cell lines MDA-MB-231 and MCF7. In MDA-MB-231 cells, a synergistic antiproliferative effect was observed with a combination of 10 µM decitabine and these three anticancer drugs, while in MCF7 cells, a semi-additive effect was observed. In a clonogenic assay, decitabine and taxotere in combination produced a greater loss of clonogenicity than either agent alone in MDA-MB-231 cells. Decitabine was found to enhance the proapoptotic effect of cisplatin on TNBC cell lines that are less sensitive to cisplatin, indicating the potential for combination therapy in TNBC. Decitabine induced the expression of apoptotic regulators and, among them, NOXA was functionally involved in decitabine-induced apoptosis.
Gene hypermethylation is important in suppressing an immune reaction to breast cancer cells
In an investigation of integrative expression and methylation of 63 cancer cell lines (breast, colorectal, and ovarian) after treatment with azaC, significant enrichment was found for immunomodulatory pathways in all three cancers (14.4-31.3%) including interferon signaling, antigen processing and presentation, and cytokines or chemokines. Strong upregulation of cancer testis antigens (CTAs) was also observed.
In an orthotopic animal models of TNBC, TNBC-intrinsic MYC impaired T cell infiltration and cytolytic function. In vitro molecular studies showed that MYC transcriptionally upregulated DNMT1, thus repressing the type I IFN response via epigenetic suppression of the stimulator of interferon genes (STING). Decitabine could reverse non-inflamed tumors into inflamed TNBC tumors and combination treatment with PD-1 inhibitor effectively reduced tumor growth in MYC-overexpressing TNBC in vivo.
DNA methyltransferase inhibitors have shown the potential to enhance the effectiveness of adoptive immunotherapy
The influence of decitabine on the immune response has led to studies on the combination of DNMTIs with immunotherapies, helping to overcome the immune escape or resistance to immunotherapy by affecting both tumor cells and the tumor microenvironment. This strategy might also be useful in chimeric antigenic receptor-T (CAR-T) cell based therapies, as myeloid derived suppressor cells (MDSC) induced immune suppressive environment is a major obstacle to the normal functioning of CAR-T cells in BC.
Mechanisms to enhance the CTL (CD8-T) response include the upregulation of tumor antigen expression, increased MHC class I expression, and blunting of MDSC expansion. In a study investigating the effect of combining decitabine, with adoptive CTL immunotherapy in the murine 4T1 mammary carcinoma model, expression of ERBB2, MHC class I molecules, and several CTAs was increased by decitabine treatment of 4T1 cells in vitro. Decitabine-treated 4T1 cells stimulated greater interferon-γ release from tumor-sensitized lymphocytes, implying increased immunogenicity. Decitabine treatment improved the efficacy of adoptive CTL immunotherapy in mice with established 4T1 tumors, with greater inhibition of tumor growth and an increased cure rate. Another study confirmed the expression of the CTA NY-ESO-1 by decitabine. Antigen expression in the MCF7 cell line was significantly increased after three days of decitabine treatment. The efficient recognition of decitabine treated BC cells by the HLA-A2 restricted NY-ESO-1 specific CAR-T cells confirmed that the de novo synthesized NY-ESO-1 antigen is functionally processed and presented.
Summary of iMN013 in vitro data
iMN013 (previously NUC013) is unique in being both a DNMTI and a ribonucleotide reductase inhibitor (RNRI). iMN013 is comprised of the base of decitabine and 5-azaC, and the difluorinated sugar of gemcitabine. This sugar results in the inhibition of ribonucleotide reductase (RNR). RNR is an enzyme that converts ribonucleoside diphosphate to deoxyribonucleoside diphosphate. In a custom siRNA screen used to identify targets that were critical for growth of TNBC cells, RNR1 and RNR2 were found to be targets for TNBC cell survival. An effect of RNR inhibition is to decrease the endogenous pools of deoxyribonucleotides, favoring the enzymatic uptake of the therapeutic deoxyribonucleotide, a phenomenon which has been described as self-potentiation. Gemcitabine is the most widely used RNR inhibitor and is approved in combination with paclitaxel for the first-line treatment of patients with mBC. As will be detailed below, combination of DNMT inhibition with RNR inhibition results in a unique therapeutic profile for iMN013 that differs in significant ways from both decitabine and gemcitabine.
The tumor suppressor gene TP53 is of vital importance in preventing human cancer development and progression. The functions and stability of the p53 protein are often abrogated via posttranslational mechanisms, such as DNA methylation, in the human cancers that harbor TP53 wild type (WT). In the Carolina Breast Cancer Study which analyzed 496 cases of invasive BC, compared with luminal A, basal-like tumors had more TP53 mutations (44% vs 15%, P < 0.001), suggesting that the majority of invasive BC tumors are TP53 WT even if TNBC. p53 is often inactivated in cancer because it can trigger cell growth arrest, apoptosis, autophagy or senescence, which are detrimental to cancer cells, and it impedes cell migration, metabolism or angiogenesis, which are favorable to cancer cell progression and metastasis. iMN013 has been shown to inhibit the growth of at least one tumor cell line from all solid tumor tissues tested in the NCI 60 cell line panel, including breast, colon, central nervous system, renal, lung, melanoma, ovarian and prostate. In this panel, iMN013 was more active than decitabine against p53-null/mutant cancer cell lines (p = 0.027) but even more so against p53 WT cell lines (p = 0.0025). This difference in activity is likely mediated by inhibition of p53R2 by iMN013. p53R2 is the p53 inducible subunit of RNR. Experimental data have shown that decitabine unexpectedly induced more apoptosis in TP53 null cells than in TP53 WT cells. These data are compatible with a clinical trial of decitabine and carboplatin where of 9 of the BC patients had available TP53 sequencing results. The 6 patients with TP53 mutations had higher objective response rate (3/6 vs. 0/3) and better overall survival (16.0 vs. 4.0 months) than the patients with TP53 WT.
iMN013 has been tested for activity in multiple cell lines. The IC50 of iMN013 in murine mammary carcinoma cell lines EMT-6 and 4T1, as well as human BC cell lines DU4475 and MCF-7, is in the nM range. The DU4475 cell line originates from a patient with TNBC.
iMN041 as an immunotherapeutic
Because of the lack of therapeutic options, there is considerable interest in immunotherapy for TNBC. Compared to BC, TNBC exhibits a greater presence of infiltrating lymphocytes, thereby establishing a favorable immune microenvironment. TNBC also demonstrates a relatively substantial tumor mutation load, providing an antigenic foundation for immune cell recognition.
Like all nucleosides with 5-aza-cytosine bases, iMN013 is subject to hydrolytic cleavage and deamination. To improve the pharmacokinetics and pharmacodynamics of iMN013 a prodrug was developed, iMN041 (previously NUC041). Based on the observation of tumor inflammation and ulceration in mice implanted with the NSCLC cancer cell line NCI-H460 treated with iMN041, it was hypothesized that the treated nude mice demonstrated enhanced NK cell activity. To further characterize the immunomodulatory effects of iMN041, iMN041 was tested in a Renca syngeneic model to determine the antitumor immune response in immunocompetent mice. When the tumor reached an approximate volume of 270 mm3, treatment of BALB/c female mice was initiated with 1 mg iMN041 SC every other day for two doses or the same volume of saline. Tumors were harvested and the cell suspension was stained with the appropriate fluorophore-conjugated monoclonal antibodies of surface markers and processed using a flow cytometer.
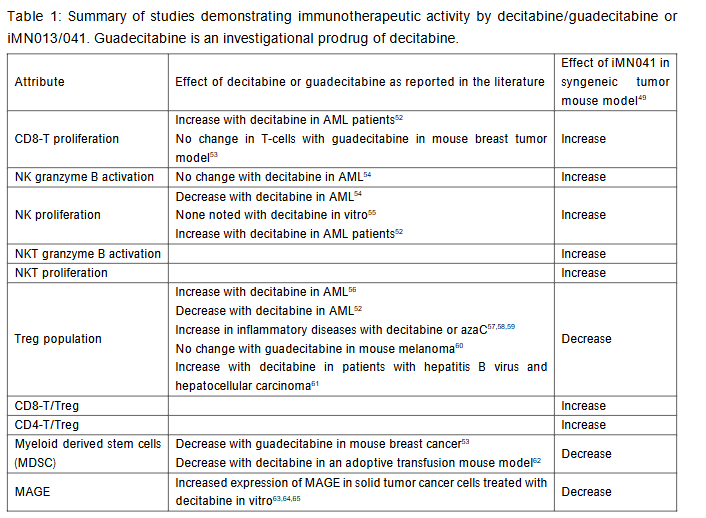
iMN041 was shown to have a unique immunotherapeutic profile. iMN041 significantly increases granzyme B in NK and NKT cells, increasing NK and NKT tumor cell cytotoxicity, and significantly increases CD8-T/Treg and CD4-T/Treg, compatible with an increase the antitumor immune response. Consistent with these data, trends in increased numbers of NK, NKT and CD8-T cells and decreased numbers of Treg cells were noted in iMN041 treated animals. MDSC were significantly decreased, suggesting a decrease in the likelihood of tumor immune escape.
As shown in the above table, data on immunomodulation by decitabine are not always consistent from study to study. It is noteworthy though, that the preponderance of data support an increase in the population of Treg cells. Indeed, decitabine has been proposed as a treatment for various inflammatory disorders. In a review of 35 preclinical studies, azaC and decitabine not only seem to be able to alleviate a number of inflammatory disorders, but also prevent solid organ rejection and graft vs. host disease in animal models. AzaC and decitabine are known to upregulate FOXP3, a master transcription factor for Treg. Seventeen studies described the effect on Treg, of which 16 studies showed an increase in Treg. Treg induce a dysfunctional state in tumor-infiltrating CTL that resembles T cell exhaustion and is characterized by low expression of effector cytokines and inefficient cytotoxic granule release. TNBC is extremely prone to drug resistance and early recurrence because of Treg infiltration into the TME. The enrichment of Treg in the TME diminishes the effects of chemotherapy and radiotherapy by creating an immunosuppressive environment through the activation of immune-inhibitory and protumor signaling. In contradistinction, administration of iMN041 resulted in a decrease in Treg leading to a significant increase in CD8-T/Treg and CD4-T/Treg.
Gemcitabine has been shown to decrease Treg cells in patients with pancreatic cancer, most likely mediated by inhibition of the small subunit of RNR, RRM2. Overexpression of the RRM2 creates an immunosuppressive tumor microenvironment. Infiltration of anti-tumor immune cells, such as CD8-T, was significantly lower in the RRM2-high group than in the RRM2-low group, whereas immunosuppressive Treg cells were more abundant in RRM2-high tumors. An elusive goal of cancer immunotherapy has been to decrease Treg cells while sparing tumor-specific effector T-cells. The difficulty of developing such therapeutics can be illustrated by the experience with bempegaldesleukin, which aimed to activate the IL-2b receptor to preferentially expand effector T cells more than Treg cells. Bempegaldesleukin demonstrated toxicity and failed to meet its primary endpoint in a Phase 3 trial in advanced melanoma.
MAGE-A, (melanoma-associated antigen)-A, is a protein that is not expressed in most normal tissues, except the testis, but is activated in a number of different cancers. MAGE-A antigen defines a very aggressive subgroup of TNBC, particularly in the absence of immune infiltration in the TME. DNMTIs have been demonstrated to increase MAGE-A levels in vitro in several tumors, including BC. Because of this activity, attempts have been made to combine decitabine with dendritic cell vaccination or T-cell therapy. iMN041 has been demonstrated to significantly decrease the proportion of cells expressing MAGE A in vivo, probably as a result of CD8-T cells targeting MAGE-A expressing tumor cells, whereas activity of CD8-T cells may be limited by Treg treated with other DNMTIs.
Efficacy of iMN041 in a mouse xenograft model of triple negative breast cancer
In an animal model of colon cancer (LoVo), iMN013 demonstrated significant suppression of tumor growth and a survival benefit compared to saline controls and decitabine. iMN041 demonstrated suppression of tumor growth, and where possible based on the number of events, survival, compared to saline control animals in models of non-small cell lung cancer (NSCLC) (NCI-H460), pancreatic cancer (SW1990 and CFPAC), renal cancer (786-O and Caki-1) and TNBC (DU4475).
In the case of DU4475, BALB/c nude female mice were inoculated subcutaneously (SC) in the right flank with tumor cells (5 X 10^6) in 200 µL PBS mixed with Matrigel (50:50) for tumor development. The animals were randomized and treatment was initiated when the average tumor size reached 60 mm3. All groups were comparable at baseline. After initiation of treatment, animals were monitored daily for morbidity and mortality. Mice were assigned to one of three groups: 0.6 mg iMN041 SC every other day, 0.8 mg iMN041 SC every other day and saline control SC every other day (n = 10/group). The treated mice had their dose held if the weight loss was > 10%. Treated mice implanted with DU4475 were initially administered iMN041 at these two different doses but mice, particularly in the 0.8 mg group, required dose withholding and adjustments; hence, the dose was decreased in both treated groups to 0.5 mg every other day on study day 8 because of concern that weight loss and drug withholding in the treated groups would complicate the assessment of drug efficacy. Tumor volume in treated mice was significantly smaller than that of control mice: In the 0.8 mg group, on days 4-20 (p < 0.05) and in the 0.6 mg group, on days 4-20 (p < 0.01). The median survival of 0.8 mg treated mice vs. control mice was 21 vs. 18 days (p = 0.019) and for the 0.6 mg treated mice was 25 vs. 18 days (p = 0.0032).
The DU4475 xenograft is a model of aggressive TNBC and there are few reports in the literature of successful treatment. Despite issues related to optimal dosing, iMN041 significantly inhibited tumor growth and improved survival. For reasons that remain undetermined mice inoculated with DU4475 were unable to tolerate the 0.8 mg every other day dose that was successfully administered in murine xenografts of renal or pancreatic cancer. Mice treated with the lower 0.6 mg dose had better outcomes because they were more likely to be treated on schedule. These data suggest that if the study had been initiated at a dose of 0.5 mg every other day, further improvements in outcome would have been observed.
Conclusions
Treatment of TNBC with a DNMTI is of interest because of the poor outcomes with current treatment regimens. Hypermethylation affects many aspects of cancer development including gene expression, in particular the expression of tumor suppressor genes, and immune response. While effective for the treatment of hematologic malignancies, decitabine and azaC have had limited success for the treatment of solid tumors, including BC. iMN013 combines the DNMTI activity of the 5-azacytosine base with the RNRI activity of gemcitabine into a novel molecule. The prodrug of iMN013, iMN041, has shown activity in multiple solid tumor models, including a model of TNBC. Decitabine has been shown to be more active against TP null cells than TP53 WT, while iMN013 has been shown to be globally more active than decitabine, but particularly so against TP53 WT tumors. The preponderance of evidence is that decitabine stimulates an increase in Treg cells, while iMN041 inhibits the proliferation of Treg. Treg cells mediate an immunosuppressive environment in tumors. Treatment with iMN041 also results in depletion of tumor cells expressing MAGE-A, which is commonly expressed in TNBC, suggesting that MAGE-A is a target of an immune system activated by iMN041. Hence, iMN041, acting through multiple pathways, may be a promising addition to the treatment armamentarium against TNBC.
Conflict of Interest:
The author is the inventor and patent holder of iMN013 and iMN041.
Funding Statement:
None.
Acknowledgements:
None.
References:
- CDC. gov. www.cdc.gov/united-states-cancer-statistics/publications/breast-cancer-stat-bite.html. Accessed 16 January 2025.
- Waks AG, Winer EP Breast cancer treatment: a review. JAMA. 2019;321(3):288-300.
- Zeichner SB, Terawaki H, Gogineni K. A review of systemic treatment in metastatic triple-negative breast cancer. Breast Cancer (Auckl). 2016;10:25-36.
- Yin L, Duan JJ, Bian XW, Yu SC. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020 Jun 9;22(1):61. doi: 10.1186/s13058-020-01296-5. PMID: 3251773; PMCID: PMC7285581.
- Zhang J, Xia Y, Zhou X et al. Current landscape of personalized clinical treatments for triple-negative breast cancer. Front Pharmacol. 2022 Sep 16;13: 977660. doi: 10.3389/fphar.2022.977660. PMID: 36188535; PMCID: PMC9523914.
- Li Y, Zhang H, Merkher Y et al. Recent advances in therapeutic strategies for triple-negative breast cancer. J Hematol Oncol. 2022 Aug 29;15(1):121. doi: 10.1186/s13045-022-01341-0. PMID: 36038913; PMCID: PMC9422136.
- Yu J, Qin B, Moyer AM et al. DNA methyltransferase expression in triple-negative breast cancer predicts sensitivity to decitabine. J Clin Invest. 2018;128(6):2376-2388.
- Connolly RM, Li H, Jankowitz RC et al. Combination epigenetic therapy in advanced breast cancer with 5-azacitidine and entinostat: a phase II National Cancer Institute/Stand Up to Cancer Study. Clin Cancer Res. 2017;23(11):2691-2701.
- Liu K, Wang YF, Cantemir C, Muller MT. Endogenous assays of DNA methyl transferases: Evidence for differential activities of DNMT1, DNMT2, and DNMT3 in mammalian cells in vivo. Mol Cell Biol. 2003;23(8):2709-19.
- Virani S, Colacino JA, Kim JH, Rozek LS. Cancer epigenetics: a brief review. ILAR J. 2012;53(3-4):359-69.
- Momparler RL. Epigenetic therapy of cancer with 5-aza- -deoxycytidine (decitabine). Semin Oncol. 2005;32:443-451.
- Daifuku R. Pharmacoepigenetics of novel nucleoside DNA methyltransferase inhibitors. In: Pharmacoepigenetics, 2nd ed. Elsevier. Cacabellos R (Ed.). 2025; in press.
- Highlights of Prescribing Information. Vidaza (Azacytidine for injection). Celgene. https://media.celgene.com/content/uploads/vidaza-pi.pdf. Accessed 10 March 2019.
- Highlights of Prescribing Information. Dacogen® (Decitabine) for Injection. Otsuka America Pharmaceutical Inc. https://www.otsukaus.com/media/static/DACOGEN-PI.pdf. Accessed 10 March 2019.
- The European Medicines Agency. Dacogen (Decitabine), 2016. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002221/human_med_001589.jsp&mid=WC0b01ac058001d124. Accessed 19 August 2018.
- Linnekamp JF, Butter R, Spijker R et al. Clinical and biological effects of demethylating agents on solid tumors – A systematic review. Cancer Treat Rev. 2017;54:10-23.
- Falahi F, van Kruchten M, Martinet N et al. Current and upcoming approaches to exploit the reversibility of epigenetic mutations in breast cancer. Breast Cancer Res. 2014 Jul 29;16(4):412. doi: 10.1186/s13058-014-0412-z. PMID: 25410383; PMCID: PMC4303227.
- Roll JD, Rivenbark AG, Sandhu R et al. Dysregulation of the epigenome in triple-negative breast cancers: basal-like and claudin-low breast cancers express aberrant DNA hypermethylation. Exp Mol Pathol. 2013;95(3):276-87.
- Wong KK. DNMT1: A key drug target in triple-negative breast cancer. Semin Cancer Biol. 2021 Jul;72:198-213. doi: 10.1016/j.semcancer.2020.05.010. Epub 2020 May 24. PMID: 32461152.
- Yang X, Phillips DL, Ferguson AT et al. Synergistic activation of functional estrogen receptor (ER)-alpha by DNA methyltransferase and histone deacetylase inhibition in human ER-alpha-negative breast cancer cells. Cancer Res. 2001 Oct 1;61(19):7025-9. PMID: 11585728.
- Man X, Li Q, Wang B et al. DNMT3A and DNMT3B in Breast Tumorigenesis and Potential Therapy. Front Cell Dev Biol. 2022 May 10;10:916725. doi: 10.3389/fcell.2022.916725. PMID: 35620052; PMCID: PMC9127442.
- Fu X, Tan W, Song Q, Pei H, Li J. BRCA1 and Breast Cancer: Molecular Mechanisms and Therapeutic Strategies. Front Cell Dev Biol. 2022 Mar 1;10:813457. doi: 10.3389/fcell.2022.813457. PMID: 35300412; PMCID: PMC8921524.
- Stolzenburg S, Beltran AS, Swift-Scanlan T et al. Stable oncogenic silencing in vivo by programmable and targeted de novo DNA methylation in breast cancer. Oncogene. 2015 Oct;34(43):5427-35. doi: 10.1038/onc.2014.470. Epub 2015 Feb 16. PMID: 25684141; PMCID: PMC4633433.
- Li C, Xiong W, Liu X et al. Hypomethylation at non-CpG/CpG sites in the promoter of HIF-1α gene combined with enhanced H3K9Ac modification contribute to maintain higher HIF-1α expression in breast cancer. Oncogenesis. 2019 Apr 2;8(4):26. doi: 10.1038/s41389-019-0135-1. PMID: 30940798; PMCID: PMC6445832.
- Liu, K., Xie, F., Gao, A. et al. SOX2 regulates multiple malignant processes of breast cancer development through the SOX2/miR-181a-5p, miR-30e-5p/TUSC3 axis. Mol Cancer 16, 62 (2017). https://doi.org/10.1186/s12943-017-0632-9
- de Heer EC, Jalving M, Harris AL. HIFs, angiogenesis, and metabolism: elusive enemies in breast cancer. J Clin Invest. 2020 Oct 1;130(10):5074-5087. doi: 10.1172/JCI137552. PMID: 32870818; PMCID: PMC7524491.
- Borges S, Döppler HR, Storz P. A combination treatment with DNA methyltransferase inhibitors and suramin decreases invasiveness of breast cancer cells. Breast Cancer Res Treat. 2014;144(1):79-91.
- Tao K, Guo S, Chen R et al. Lysophosphatidic acid receptor 6 (LPAR6) expression and prospective signaling pathway analysis in breast cancer. Mol Diagn Ther. 2019. doi: 10.1007/s40291-019-00384-3.
- Umeh-Garcia M, O’Geen H, Simion C et al. Aberrant promoter methylation contributes to LRIG1 silencing in basal/triple-negative breast cancer. Br J Cancer. 2022 Aug;127(3):436-448. doi: 10.1038/s41416-022-01812-8. Epub 2022 Apr 19. PMID: 35440669; PMCID: PMC9346006.
- Palomeras S, Diaz-Lagares Á, Viñas G et al. Epigenetic silencing of TGFBI confers resistance to trastuzumab in human breast cancer. Breast Cancer Res. 2019 Jul 5;21(1):79. doi: 10.1186/s13058-019-1160-x. PMID: 31277676; PMCID: PMC6612099.
- Ponnusamy L, Mahalingaiah PKS, Chang YW, Singh KP. Reversal of epigenetic aberrations associated with the acquisition of doxorubicin resistance restores drug sensitivity in breast cancer cells. Eur J Pharm Sci. 2018 Oct 15;123:56-69. doi: 10.1016/j.ejps.2018.07.028. Epub 2018 Jul 19. PMID: 30016648.
- He DX, Xia YD, Gu XT, Jin J, Ma X. A transcription/translation-based gene signature predicts resistance to chemotherapy in breast cancer. J Pharm Biomed Anal. 2015;102:500-8.
- Mirza S, Sharma G, Pandya P, Ralhan R. Demethylating agent 5-aza-2-deoxycytidine enhances susceptibility of breast cancer cells to anticancer agents. Mol Cell Biochem. 2010;342(1-2):101-9.
- Hurtubise A, Momparler RL. Evaluation of antineoplastic action of 5-aza-2′-deoxycytidine (Dacogen) and docetaxel (Taxotere) on human breast, lung and prostate carcinoma cell lines. Anticancer Drugs. 2004;15(2):161-7.
- Nakajima W, Miyazaki K, Sakaguchi M et al. Epigenetic Priming with Decitabine Augments the Therapeutic Effect of Cisplatin on Triple-Negative Breast Cancer Cells through Induction of Proapoptotic Factor NOXA. Cancers (Basel). 2022 Jan 4;14(1):248. doi: 10.3390/cancers14010248. PMID: 35008411; PMCID: PMC8749981.
- Li H, Chiappinelli KB, Guzzetta AA et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget. 2014;5(3):587-98.
- Wu SY, Xiao Y, Wei JL et al. MYC suppresses STING-dependent innate immunity by transcriptionally upregulating DNMT1 in triple-negative breast cancer. J Immunother Cancer. 2021 Jul;9(7):e002528. doi: 10.1136/jitc-2021-002528. PMID: 34321275; PMCID: PMC8320259.
- Xu Y, Li P, Liu Y et al. Epi-immunotherapy for cancers: rationales of epi-drugs in combination with immunotherapy and advances in clinical trials. Cancer Commun (Lond). 2022 Jun;42(6):493-516. doi: 10.1002/cac2.12313. Epub 2022 Jun 1. PMID: 35642676; PMCID: PMC9198339.
- Corti C, Venetis K, Sajjadi E et al. CAR-T cell therapy for triple-negative breast cancer and other solid tumors: preclinical and clinical progress. Expert Opin Investig Drugs. 2022 Jun;31(6):593-605. doi: 10.1080/13543784.2022.2054326. Epub 2022 Mar 24. PMID: 35311430.
- Terracina KP, Graham LJ, Payne KK et al. DNA methyltransferase inhibition increases efficacy of adoptive cellular immunotherapy of murine breast cancer. Cancer Immunol Immunother. 2016;65(9):1061-73.
- Klar AS, Gopinadh J, Kleber S, Wadle A, Renner C. Treatment with 5-Aza-2′-deoxycytidine induces expression of NY-ESO-1 and facilitates cytotoxic T lymphocyte-mediated tumor cell killing. PLoS One. 2015;10(10):e0139221.
- Bennett CN, Tomlinson CC, Michalowski AM et al. Cross-species genomic and functional analyses identify a combination therapy using a CHK1 inhibitor and a ribonucleotide reductase inhibitor to treat triple-negative breast cancer. Breast Cancer Res. 2012;14(4):R109.
- Daifuku R, Hu Z, Saunthararajah Y. 5-aza-2′,2′-Difluoro Deoxycytidine (NUC013): A Novel Nucleoside DNA Methyl Transferase Inhibitor and Ribonucleotide Reductase Inhibitor for the Treatment of Cancer. Pharmaceuticals (Basel). 2017 Jul 20;10(3):65. doi: 10.3390/ph10030065. PMID: 28726739; PMCID: PMC5620609.
- Highlights of prescribing information. Gemzar (gemcitabine for injection). https://pi.lilly.com/us/gemzar.pdf. Accessed 19 February 2019.
- Carey LA, Perou CM, Livasy CA et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA. 2006;295(21):2492-502.
- Zhang Q, Zeng SX, Lu H. Targeting p53-MDM2-MDMX loop for cancer therapy. Subcell Biochem. 2014;85:281-319. doi: 10.1007/978-94-017-9211-0_16. PMID: 25201201; PMCID: PMC4472007.
- Nieto M, Samper E, Fraga MF, González de Buitrago G, Esteller M, Serrano M. The absence of p53 is critical for the induction of apoptosis by 5-aza-2′-deoxycytidine. Oncogene. 2004 Jan 22;23(3):735-43. doi: 10.1038/sj.onc.1207175. PMID: 14737108.
- Wang H, Wang Z, Wang Z et al. Decitabine induces IRF7-mediated immune responses in p53-mutated triple-negative breast cancer: a clinical and translational study. Front Med. 2024 Apr;18(2):357-374. doi: 10.1007/s11684-023-1016-8. Epub 2023 Dec 29. PMID: 38157193.
- Daifuku, R., Zhang, Y., Wang, J., Gu Q. iMN041 is an immunotherapeutic and an effective treatment in mouse xenograft models of pancreatic cancer, renal cancer and triple negative breast cancer. Transl Med Commun 9, 2 (2024). https://doi.org/10.1186/s41231-024-00161-3
- Liu Y, Hu Y, Xue J et al. Advances in immunotherapy for triple-negative breast cancer. Mol Cancer. 2023 Sep 2;22(1):145. doi: 10.1186/s12943-023-01850-7. Erratum in: Mol Cancer. 2023 Sep 14;22(1):154. doi: 10.1186/s12943-023-01858-z. PMID: 37660039; PMCID: PMC10474743.
- Daifuku R, Grimes S, Stackhouse M. NUC041, a Prodrug of the DNA Methytransferase Inhibitor 5-aza-2′,2′-Difluorodeoxycytidine (NUC013), Leads to Tumor Regression in a Model of Non-Small Cell Lung Cancer. Pharmaceuticals (Basel). 2018 Apr 23;11(2):36. doi: 10.3390/ph11020036. PMID: 29690576; PMCID: PMC6027359.