






Early-Onset Dementia with Lewy Bodies and PRKN Mutation
Case of Early-onset Dementia with Lewy Bodies Associated with PRKN Gene Mutation
Leila Lotfi, MD1, Neeraj Singh, MD2
- Department of Medicine, Long Island Jewish Forest Hills Hospital, Northwell Health, New York, NY, USA
- Department of Neurology, Northwell Health Neurosciences Institute, New York, NY, USA
OPEN ACCESS
PUBLISHED 31 March 2025
CITATION Lotfi, L., and Singh, N., 2025. Case of Early-onset Dementia with Lewy Bodies Associated with PRKN Gene Mutation. Medical Research Archives, [online] 13(3). https://doi.org/10.18103/mra.v13i3.6319
COPYRIGHT: © 2025 European Society of Medicine. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
ABSTRACT
Dementia with Lewy bodies is the second most common neurodegenerative form of dementia following Alzheimer’s disease, affecting 0.4% of people over the age of 65 every year. It is more commonly diagnosed in men than in women, and typically over the age of 50. It is a synucleinopathy, in which alpha-synuclein inclusions accumulate in neurons, similar to idiopathic Parkinson’s disease and multiple system atrophy. Clinical features include cognitive decline, bradykinetic movements, and visual hallucinations. Neuroimaging in dementia with Lewy bodies typically reveals occipital and parietal lobe atrophy and hypometabolism. Some cases of dementia with Lewy bodies and idiopathic Parkinson’s disease have been shown to be associated with genetic mutations, such as in the GBA gene. In this case report, we present a unique case of a young woman who started showing clinical features of dementia with Lewy bodies at the age of 44, with further confirmation of the diagnosis from neuroimaging studies. However, her genetic test results revealed the presence of a PRKN gene mutation, which has been described with Parkinson’s disease, even though she did not show typical clinical features of this disease. This report reveals a potential unique association of the PRKN mutation with early-onset dementia with Lewy bodies, indicating that we may have an incomplete understanding of the function of such genes in cognitive development, as well as the phenotypic presentations of dementia with Lewy bodies and Parkinson’s disease.
Keywords: dementia, Lewy bodies, PRKN gene mutation, early-onset dementia, neurodegenerative disorders
Introduction
Dementia with Lewy bodies (DLB) is a neurodegenerative disorder that features at least three of the following clinical features, based on criteria updated in 2017: fluctuating cognition, rapid eye movement (REM) sleep behavior disorder, at least one Parkinsonian feature, and recurrent visual hallucinations. Fluctuating cognition can affect executive function, visuospatial function, and attention early during the course of DLB. Disordered REM sleep behavior can also precede cognitive impairments in DLB. Parkinsonian features that can be seen in DLB include rigidity, bradykinetic movements, and resting tremors.
The diagnosis of DLB typically occurs between ages 51 and 90 years, with an average age of diagnosis of 75 years, and more often in men than in women. The annual incidence of DLB is 4% of new dementia cases, while the prevalence ranges between zero and 22.8% of all cases of dementia, with higher prevalences reported in patient care facilities as compared with the community. However, with advances in diagnostic tools for neurodegenerative disease, there has been a recent increase in the reported prevalence of DLB. There is a higher risk of DLB in patients with a family history of any type of dementia, although diagnosis may be delayed because some features can be confused with psychiatric symptoms. Some early-onset DLB cases have fewer amnestic features than late-onset DLB cases. Visual hallucinations and parkinsonian features in particular are helpful to distinguish early-onset dementia with Lewy bodies from early-onset Alzheimer’s dementia.
The prognosis for survival can be between 2 and 20 years from the age of diagnosis, although it is most often between 5 and 7 years from the age of diagnosis. There is no curative treatment for DLB, so treatments are administered to manage the associated psychiatric, cognitive, and motor symptoms. Acetylcholinesterase inhibitors have been shown to be effective in slowing down cognitive decline, most notably with rivastigmine and donepezil, although with a greater risk of adverse effects with rivastigmine.
Case report
We present the case of a 42-year-old woman with a medical history of Hashimoto thyroiditis, cerebral meningioma, and anxiety who presented with new visual hallucinations and head tremors. The patient also reported experiencing sensations of internal tremors, chills, nocturnal perspiration, heat sensitivity, cognitive impairment, and profound fatigue, particularly following an episode of tremors in the head. These symptoms became more frequent and evident over the course of a few months. Additional symptoms such as muscle spasms and sleep disturbances emerged, significantly impacting her quality of life. Family history was positive for transient ischemic attack in the mother, stroke, and hand tremors in the father. The patient had occasional alcohol consumption, without any history of smoking or illicit drug use. Prior to evaluation by our team, she was evaluated by multiple specialists, including a psychiatrist and a neurologist with a memory disorders subspecialty. Despite early features consistent with DLB, the symptoms were dismissed as psychiatric symptoms, and medication for anxiety was provided, which yielded only minimal improvement.
Past Medical History: Hashimoto thyroiditis, Cerebral meningioma, Anxiety
Family History: Transient ischemic attack in mother, Stroke and hand tremors in father
Medications: Levothyroxine, Fluoxetine
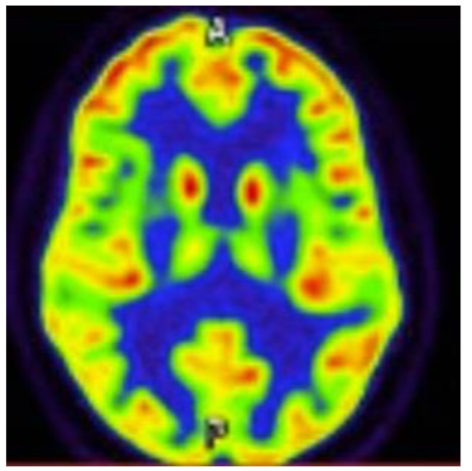
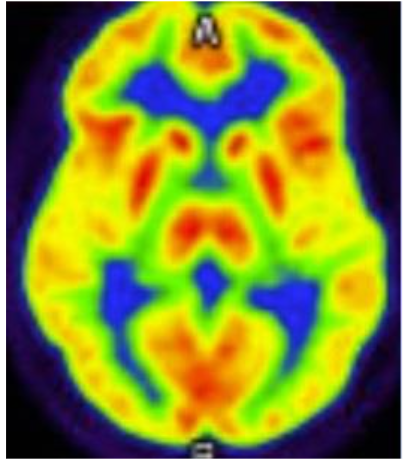
Figure 2 shows a PET-FDG MR Brain axial cross-section performed 14 months later, which re-demonstrates bilateral parietal and occipital hypometabolism, with preserved cingulate island sign, further supporting a diagnosis of dementia with Lewy bodies.
Complementary evaluations
The patient had multiple occurrences of elevated Thyroid Stimulating Hormone (TSH) levels ranging from 4.32 to 6.1, as well as an elevated Thyroid Peroxidase Antibody level of 227 IU/mL. Various laboratory tests were conducted, including total Triiodothyronine (T3), free and total thyroxine (T4), thyroglobulin antibody, complete blood count, complete metabolic panel, as well as tests for magnesium, copper, vitamin E, vitamin B1, vitamin B12, folate, methylmalonic acid, homocysteine, HbA1c, ACE, RPR, ESR, and CRP, all of which yielded normal results.
The patient’s serum antibodies for Lyme, Anti-Hu, Anti-Ri, Anti-Yo, Anti-NMDA, Anti-SSA/Anti-SSB, Anti-dsDNA, RNP, Anti-Smith, Anti-Scl-70, Antichromatin, Anti-Jo-1, Anti-Centromere, Anti-Amphiphysin, Anti-VKGC, West Nile Virus, Antigliadin, and Liver-Kidney Microsomal Ab were all negative, while the Antinuclear Antibody (ANA) had borderline titer at 1:80. Additionally, genetic panels for Hereditary Parkinson Disease, Hereditary Amyotrophic Lateral Sclerosis, Frontotemporal Dementia, and Alzheimer Disease were conducted, revealing a positive result for a heterozygous PRKN mutation in the patient.
Brain imaging studies, including PET-FDG CT and PET-FDG MR within 14 months of the CT scan, showed hypometabolism in the bilateral parietal and occipital regions with preserved cingulate island sign, supporting the diagnosis of early-onset DLB.
Discussion
This case demonstrates that DLB can be diagnosed early in life even though the reported average age of diagnosis is 75 years. Computed tomography (CT) imaging of the brain typically does not reveal structural changes, but brain magnetic resonance imaging (MRI) can reveal atrophy in the occipital and parietal lobes and preservation of the temporal lobe volumes. When a brain CT or MRI image is performed with fluorodeoxyglucose positron emission tomography (PET-FDG), it can also reveal occipital and parietal lobe hypometabolism, often with preserved metabolism in the cingulate cortex. Single-photon emission computed tomography (SPECT) scans can also reveal decreased dopamine transporter uptake in the basal ganglia in DLB, but this can also be seen with PD.
Histopathological studies of neurons affected by DLB reveal eosinophilic cytoplasmic depositions of alpha-synuclein in clusters, which are called Lewy bodies. In some cases of early-onset DLB, instead of Lewy bodies, there may be pale bodies, which are small, lightly stained aggregates of proteins within neuronal bodies. There are also losses of dopamine-producing cells in the basal ganglia and cholinergic cells in the basal forebrain. However, histochemical staining in DLB can be difficult and sometimes can be difficult to distinguish from other synucleinopathies, such as PD, so DLB has been underdiagnosed based on histopathological studies.
Genetic studies have been pursued to identify familial cases of DLB, which have typically been diagnosed in the fourth and fifth decades of life. A multicenter genomic study has identified five significant independent loci that can influence the risk for developing DLB. These include the GBA, BIN1, TMEM175, SNCA-AS1, and APOE genes. Of these genes, the highest risks of developing DLB are associated with the GBA gene, which encodes the lysosomal enzyme glucocerebrosidase; the APOE gene, which encodes apolipoprotein E; and the SNCA-AS1 gene, which encodes alpha-synuclein.
The PRKN gene encodes parkin, which is an E3 ubiquitin-protein ligase that is important for protein ubiquitination and removal of damaged mitochondria. A mutation in the PRKN gene is transmitted in an autosomal recessive fashion. This mutation is the most common cause of early-onset PD, with characteristic features of bradykinesia, rigidity, and tremors, but typically with relative preservation of cognitive function as compared with other forms of PD. The PRKN mutation has not typically been associated with DLB, which tends to have an earlier onset of cognitive impairment than all forms of PD.
Conclusion
Although DLB has a relatively advanced average age of onset, it can occur with an earlier onset especially in inherited forms. Although multiple genetic mutations have been reported with early-onset DLB, this case reveals a relationship between early-onset DLB and a mutation in the PRKN gene. While DLB and PD are both synucleinopathies, mutations in the PRKN gene have heretofore reported as common causes for early-onset PD, but not DLB. This patient’s clinical symptoms and neuroimaging, including PET-FDG Brain imaging, support the diagnosis of DLB instead of PD or AD, indicating a new association with PRKN gene mutations and development of early-onset DLB. This case demonstrates the importance of early testing for dementia for patients under the age of 50 years who show historical and clinical signs of cognitive decline and behavioral changes, especially if they have a family history of dementia, since this can lead to early diagnosis and treatment.
Conflicts of interest statement
All Authors declare no conflict of interest.
Funding statement
All Authors declare no external sources of funding.
Acknowledgments
None
References
- McKeith IG, Boeve BF, Dickson DW, et. al. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology. 2017 Jul 4;89(1):88-100.
- Boot BP, McDade EM, McGinnis SM, et. al. Treatment of dementia with lewy bodies. Curr Treat Options Neurol. 2013 Dec;15(6):738-64.
- Vann Jones SA, O’Brien JT. The prevalence and incidence of dementia with Lewy bodies: a systematic review of population and clinical studies. Psychol Med. 2014 Mar;44(4):673-83.
- Savica R, Grossardt BR, Bower JH, et. al. Incidence of dementia with Lewy bodies and Parkinson disease dementia. JAMA Neurology. 2013 Nov; 70(11):1396-402.
- Sim J, Li Huihua, Hameed S, et. al. Clinical Manifestations of Early-Onset Dementia With Lewy Bodies Compared With Late-Onset Dementia With Lewy Bodies and Early-Onset Alzheimer Disease. JAMA Neurol. 2022;79(7):702-709.
- Stinton C, McKeith I, Taylor JP, et. al. Pharmacological Management of Lewy Body Dementia: A Systematic Review and Meta-Analysis. Am J Psychiatry. 2015 Aug 1;172(8):731-42.
- Yousaf T, Dervenoulas G, Valkimadi PE, et. al. Neuroimaging in Lewy body dementia. J Neurol. 2019 Jan;266(1):1-26.
- Gomperts SN. Lewy Body Dementias: Dementia With Lewy Bodies and Parkinson Disease Dementia. Continuum (Minneap Minn). 2016 Apr;22(2 Dementia):435-63.
- Wakabayashi K, Tanji K, Odagiri S, et. al. The Lewy body in Parkinson’s disease and related neurodegenerative disorders. Mol Neurobiol. 2013 Apr;47(2):495-508.
- Chia R, Sabir MS, Bandres-Ciga S, et. al. Genome sequencing analysis identifies new loci associated with Lewy body dementia and provides insights into its genetic architecture. Nat Genet. 2021 Mar;53(3):294-303.
- Clausen L, Okarmus J, Voutsinos V, et. al. PRKN-linked familial Parkinson’s disease: cellular and molecular mechanisms of disease-linked variants. Cell Mol Life Sci. 2024 May 20;81(1):223.
- Cherian A, Divya KP, Vijayaraghan A. Parkinson’s disease – genetic cause. Curr Opin Neurol. 2023 Aug 1;36(4):292-301.
- Daida K, Funayama M, Billingsley KJ, et. al. Long-Read Sequencing Resolves a Complex Structural Variant in PRKN Parkinson’s Disease. Mov Disord. 2023 Dec;38(12):2249-2257.