





Endometriosis and Ovarian Cancer: Unraveling the Link
Endometriosis-Related Ovarian Carcinogenesis: Unravelling The Roots of a Long-Standing Enigma
Demetrio Larraín, MD¹, Nicanor Barrena, MD²
- Minimally Invasive Gynecologic Surgery and Endometriosis Unit; Obstetrics and Gynecology Department, Clínica Santa María, Santiago, Chile
- Gynecologic Oncology Unit; Obstetrics and Gynecology Department, Clínica Santa María, Santiago, Chile
OPEN ACCESS
PUBLISHED: 30 November 2024
CITATION: Larraín, D. and Barrena, N., 2024. ENDOMETRIOSIS-RELATED OVARIAN CARCINOGENESIS: UNRAVELLING THE ROOTS OF A LONG-STANDING ENIGMA.
Medical Research Archives, [online]12(11). https://doi.org/10.18103/mra.v1 2i11.5965
COPYRIGHT: © 2024 European Society of Medicine. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
DOI https://doi.org/10.18103/mra.v1 2i11.5965
ISSN 2375-1924
ABSTRACT
Endometriosis is a gynecologic disease in which ectopic endometrial tissue causes both chronic pelvic pain and infertility. Likewise, this entity has been linked with the development of certain types of epithelial ovarian cancer. The mechanisms underlying this association have remained largely elusive yet recent advances in terms of identifying histologically well-defined precursor lesions as well as key molecular and genetic abnormalities involved in the endometriosis malignant transformation have dramatically improved the understanding of this clinical conundrum.
Introduction
Endometriosis is a chronic benign estrogen-dependent inflammatory disease, defined by the presence of functional endometrial tissue (glands and stroma) outside of the uterine cavity¹. In addition to chronic pain and infertility, endometriosis has been associated with an increased risk of certain cancers including epithelial ovarian cancer, especially clear cell and endometrioid carcinomas²³.
Despite both endometriosis and ovarian cancer share several characteristics, such as risk factors, clonal growth, genetic alterations, immune dysregulations, estrogen dependency, decreased apoptosis, angiogenesis, invasion and metastatic potential, endometriosis is a frequent condition affecting 10–15% women of reproductive age¹, but the lifetime risk for developing ovarian cancer in general population is only 1.3% (one in 78 women)⁴. This lifetime risk reaches 2.5% in patients with endometriosis³. A large prospective cohort study in which 6398 women with a clinically documented ovarian endometrioma were followed for up to 17 years, demonstrated that 0.72% of endometriomas undergo malignant transformation⁵.
In general, it has been estimated that the risk of neoplastic transformation of endometriosis is about 0.5–1% cases⁶. Given such low rate of malignant transformation, endometriosis should not be considered as a premalignant lesion, but a disease with the potential to develop malignancy. However, some studies have identified risk factors for ovarian cancer development in patients with endometriosis, such as diagnosis of endometriosis after the age of 45 years, endometrioma size > 9 cm and/or complex aspect on ultrasound⁸⁹.
Despite a substantial body of evidence suggests a link between endometriosis and epithelial ovarian cancer, the plausible underlying mechanism remains elusive. In recent years, many advances have been made in the understanding of genetics and molecular mechanisms underlying endometriosis-associated ovarian carcinogenesis, and aberrations in several complex cell signaling pathways have been shown to be involved in the process. Despite none of such alterations alone have proven to be enough for tumoral development, most of these altered signaling cascades converge in a summative way, making the understanding of endometriosis-associated ovarian carcinogenesis even more complex.
In this narrative review we focus on integrating this new knowledge and the current theories regarding endometriosis-associated ovarian carcinogenesis.
Endometriosis and epithelial ovarian cancer types: Epidemiologic studies
Epidemiological evidence from large studies has demonstrated that endometriosis is an independent risk factor for ovarian cancer, particularly clear cell and endometrioid subtypes³¹⁰¹². In a pooled analysis of 13 case-control studies, the Ovarian Cancer Association Consortium reported a greater risk of both clear cell (Odds Ratio (OR) 3.05 [95% CI, 2.43–3.84]) and endometrioid (OR 2.04 [95% CI, 1.67–2.48]) ovarian carcinomas¹¹. However, the presence of endometriosis has been also associated with other ovarian cancer types, such as low-grade serous carcinoma and carcinosarcoma³¹¹¹³. Association of endometriosis with high-grade serous or mucinous ovarian carcinomas have been less consistent¹³.
Recently, a large population-based study confirmed a significantly higher incidence of clear cell and endometrioid ovarian cancer among women with histologically proven endometriosis¹².
Endometriosis typology and ovarian cancer risk
Endometriosis is classically divided in three clinical forms: peritoneal, ovarian (endometrioma) and deep endometriosis. Despite the higher risk of ovarian cancer among endometriosis patients, only a few studies have evaluated the risk of ovarian cancer according endometriosis typology.
A recent large population-based study of 78,893 endometriosis patients reported the highest ovarian cancer risk among women with deep endometriosis and/or ovarian endometriomas (adjusted Hazard Ratio (aHR) 9.66 [95% CI, 7.77–12]) for all epithelial ovarian cancers compared with individuals without endometriosis. This risk remained elevated during the first 5 years from the diagnosis and after 20 years of follow-up, following a U-shaped relationship between endometriosis and ovarian cancer across follow-up time¹³.
Saavalainen et al¹⁴ assessed the risk of gynecologic cancer in a population-based study including 49,933 patients with surgically verified endometriosis according endometriosis type. The authors reported an increased risk of ovarian cancer in patients with ovarian endometriosis, especially endometrioid (Standardized Incidence Ratio (SIR) 4.72 [95% CI, 2.75–7.56]), and clear cell (SIR 10.1 [95% CI, 5.50–16.9]) subtypes. The increased risk started from 5 years after surgery and from the age of 30 years onwards. In addition, peritoneal endometriosis was associated with a slightly increased risk for endometrioid ovarian cancer (SIR 2.03 [95% CI, 1.05–3.54]) and with clear cell histology after 10 years of follow-up (SIR 3.79 [95% CI, 1.39–8.24]). There was no association between deep endometriosis and the risk of ovarian cancer; however, there were only 3 patients with deep endometriosis in the cohort¹⁴. Interestingly, the risk of borderline ovarian tumors was significantly elevated during the first 6 months after surgery in patients with ovarian endometriosis¹⁴.
Endometriosis-associated ovarian cancer: a distinct clinical entity?
Previous epidemiological data have shown that EAOC has a different biological behavior compared to ovarian cancer occurring in the absence of endometriosis (non-EAOC), suggesting that it may represent a different clinical entity with a more favorable prognosis¹⁰¹⁵¹⁶.
Wang et al¹⁷ found that compared with non-EAOC, patients with EAOC were proved to be younger (p=0.03) and more likely to be premenopausal at diagnosis (p=0.005). Furthermore, EAOC patients were more likely to present lower or normal CA125 preoperative levels, to be diagnosed at earlier stage than non-EAOC (88.2% vs. 15.8% at Stage I) and to have a significant overrepresentation of endometrioid and clear cell subtypes compared to non-EAOC.
In 2018, Bassiouny et al¹⁶ investigated the clinical-pathologic characteristics and outcome of EAOC compared with non-EAOC in a large cohort. Patients with EAOC were younger at presentation, presented at an earlier stage and had lower recurrence rate compared with non-EAOC patients (26.8% vs. 45.3%, respectively; p<0.001). In addition, EAOC patients had longer estimate of 5-year disease-free survival compared with non-EAOC women (70% vs. 39.3%, respectively; p<0.001).
In the same line, other studies also reported a significantly better overall survival in patients with EAOC compared to non-EAOC patients¹⁸¹⁹. Better prognosis associated to EAOCs could be explained by the higher prevalence of early-stage and low-grade tumors when compared with non-EAOC¹⁸.
Conversely, other authors have reported conflicting results¹⁵²⁰. Davis et al²⁰ reported that despite EAOC presented lower recurrence rate and improved 5-year disease-free survival than non-EAOC, this did not translate into a difference in OS.
The association between the presence of endometriosis and survival is less consistent since after controlling confounding factors, such as stage, no association emerged between the presence of endometriosis and survival²¹²³.
Noteworthy, a distinctive characteristic of EAOC is the well documented increased incidence of concurrent malignancy at the time of diagnosis. Davis et al²⁰ found that 23.8% of EAOC had a concurrent primary cancer and that 94.1% were endometrial cancer. This finding was confirmed by Mangili et al¹⁵ that reported that 33% of patients with endometriosis-associated ovarian endometrioid cancer have a diagnosis of endometrial hyperplasia, and 33% of these women had a concomitant endometrial carcinoma. Among endometrial cancer patients, 92% had the same histology and grade in both the ovarian and uterine malignancy. The histological and clinical parallelism between EAOC, in particular the endometrioid subtype, and synchronous endometrial cancers had led to much debate over the molecular mechanisms that dictate the malignant transformation of endometriosis.
Thus, there is enough evidence to suggest that when the diagnosis of an EAOC is faced, particularly endometrioid, it should prompt additional study to exclude a synchronous primary endometrial carcinoma.
Atypical endometriosis and ovarian cancer risk
The possible association between endometriosis and ovarian cancer was first established in 1925 by Sampson²⁴. Later on, in 1953, Scott added the histological demonstration of benign endometriotic lesions adjacent to malignant tissue as a fourth criteria²⁵. Additionally, in 2004, Van Gorp et al¹⁰ proposed a classification for all cases of ovarian with concomitant endometriosis. These criteria are still used in the pathological diagnosis of endometriosis-associated ovarian cancer (EAOC) and are summarized in Table 1.
Table 1. Criteria for diagnosis and classification of endometrial-associated ovarian cancer
a) Sampson and Scott’s criteria²⁴²⁵
- Evidence of coexisting tumor and endometriosis in the same ovarian location
- Exclusion of a secondary malignancy elsewhere
- Histological pattern that resembles endometrial origin
- Histological demonstration of benign lesions of endometriosis adjacent to malignant tissue
b) Van Gorp’s criteria¹⁰
A) Ovarian cancers with histological proof of transition from ovarian endometriosis to ovarian cancer according to the definition of Sampson and Scott.
B) Ovarian cancers with endometriosis in the same ovary but without histological proof of transition or without knowledge whether this transition was further investigated or not.
C) Ovarian cancers with concomitant endometriosis at any location in the pelvis: endometriosis in the bilateral or contralateral ovary, extragonadal endometriosis, or without specification about lateralization and/or localization of the lesion.
Although the processes underlying the development of ovarian carcinoma from benign endometriosis are not fully understood, several authors have suggested that malignant transformation may be associated with the presence of atypical endometriosis²⁶. Atypical endometriosis may arise in endometriotic tissue under the stimulation of chronic inflammatory processes and oxidative stress, and it is widely accepted that it represents an intermediary entity (precursor lesion) between endometriosis and EAOC²⁶. Indeed, several studies have demonstrated both the spatial (continuous transition from benign epithelium through atypical endometriosis to carcinoma) and chronological relationship between atypical endometriosis and ovarian cancer¹⁰²⁶²⁷. Reported rates of atypical endometriosis vary from 20–80% in patients with EAOC. Reasons for such variations is that there is a lack of agreement on pathologic criteria for the diagnosis of atypical endometriosis²⁷²⁸. In a recent systematic review, the prevalence of atypical endometriosis was significantly higher among patients with EAOC (22.8–34.6%) compared to those with endometriosis without malignancy (<1–5.8%)²⁶. Despite atypical endometriosis may be seen relatively frequently associated with EAOC, it is an uncommon diagnosis reported only in 2–3% of endometriomas²⁸. Hence, these data suggest that endometriosis rarely transform to carcinoma, occasionally passing through an intermediate lesion, namely, atypical endometriosis.
The term atypical endometriosis includes two different histologic findings: 1) cytological or cellular atypia which corresponds to the presence of atypia within the epithelial lining on endometrial cysts. However, this finding is usually found in endometriomas with abundant fibrosis and inflammation and could correspond to a reactive change secondary to repeated hemorrhagic episodes²⁷; and 2) architectural atypia or hyperplasia, which seems to replicate the premalignant nature of atypical endometrial hyperplasia with respect to endometrial cancer (simple or complex, with or without cytologic atypia)²⁶²⁹.
Traditionally, both cytologic and architectural atypia were considered as a single entity. However, some authors have suggested that they must be considered as different subtypes of atypical endometriosis since their clinical significance and prognostic implications are different²⁶. While cytologic atypia can be observed in 11.1% of EAOC, the presence of architectural atypia has been reported in up to 88.9% of these patients (<0.009)⁶. Since architectural atypical endometriosis carries a higher risk of conversion to ovarian cancer, some authors have applied the term borderline endometrioid tumor for cases of endometriosis where foci resembling atypical hyperplasia are found⁷.
To date, most studies on atypical endometriosis have been performed using mixed criteria (architectural and cytologic atypia) for the diagnosis of atypical endometriosis and only few papers have considered the definition of architectural atypical endometriosis, distinguishing its significance compared to cytologic atypia. In a recent retrospective study with over an 11-year follow-up, Wepy et al³⁰ found synchronous or subsequent tubo-ovarian neoplasia in 25% of atypical endometriosis cases (9 of 24 patients), with architectural atypia being the most significant alteration in patients with synchronous/subsequent neoplasia. Despite these studies are small, they provide some evidence to suggest that the presence of architectural atypical endometriosis should prompt additional study to exclude a concurrent neoplasia.
Interestingly, histological evidence of both squamous and mucinous endometrioid metaplasia was observed more frequently in association with endometrioid ovarian cancer arising from endometriosis than in non-EAOC, thus supporting the concomitant coexistence of precancerous stage areas¹⁵²⁷. Furthermore, the presence of mucinous metaplasia has been found to be an independent predictor for the detection of endometriosis within endometrioid ovarian cancer¹⁵. However, endometriotic metaplasia is a common finding in both endometriosis and EAOC and its association with cancer is unclear³⁰³¹.
The evidence of a continuum transition from endometriosis to EAOC comes studies different immunohistochemical and molecular markers. A potential marker indicative of premalignant potential in atypical endometriosis is the expression of Ki-67. High Ki-67 expression is associated with increased mitotic activity and aggressive tumor behavior. Ñiguez Sevilla et al⁶ reported higher Ki-67 proliferative index in patients with atypical endometriosis than in those with typical endometriosis (p<0.001). Ki-67 index was also higher in patients with architectural atypical endometriosis compared to those with cytologic atypia (p<0.004). Similar results have been reported by other studies³²³³.
As chronic inflammation may have a role in the pathogenesis of atypical endometriosis (reactive changes)²⁶, some studies have evaluated inflammatory markers such as COX-2 expression in endometriosis⁶, suggesting a higher COX-2 positivity in typical compared to atypical endometriosis. Despite COX-2 positivity was higher in atypical endometriosis with cytologic atypia (80%) compared to architectural type (20%), the difference was not significant (p<0.09). These findings support the reactive nature of cytological atypia.
In 2013, Vercellini et al³⁴ evaluated the oncofetal protein IMP3 as a potential immunohistochemical marker of atypical endometriosis in a retrospective analysis of 874 endometriomas. Their results suggest that immunohistochemical IMP3 expression is a simple, inexpensive, and sensitive test that can be useful clinical practice to triage tool for atypical endometriosis and confounding benign conditions.
In a recent study, Del Mundo²⁹ et al showed the potential diagnostic utility of a 3-immunohistochemical marker panel used in the diagnosis of endometrial pathology (β-catenin, PAX2, PTEN), in the characterization of endometrioid ovarian lesions, including endometriosis, endometriosis with cytologic atypia, atypical endometriosis with architectural atypia (defined as endometrioid borderline tumors) and endometrioid ovarian cancer. The incidence of aberrancy for the 3 markers increased along the histologic spectrum of neoplastic progression, suggesting that this 3-marker panel could be useful in identifying atypical endometriosis at higher risk of neoplastic progression.
Mechanisms involved in endometriosis-related ovarian carcinogenesis
Current research supports the idea that, until now, epithelial ovarian cancer has been erroneously regarded as a single disease. Recent morphologic, immunohistochemical and molecular genetic studies have unexpectedly challenged our understanding about the origins of ovarian cancer and nowadays a new paradigm for ovarian carcinogenesis, based on a dualistic model, is widely accepted. In this model, which highlights the heterogeneity of ovarian carcinoma, epithelial tumors are divided into two broad categories designated as type I and type II³⁵³⁶. Both groups of tumor have marked morphological and genetic differences (Table 2), suggesting that different types of ovarian carcinomas develop along different molecular pathways³⁵³⁷. Moreover, most of what seemed to be primary ovarian cancer, namely serous, endometrioid and clear cell carcinomas would arise from fallopian tube and endometrium (Müllerian-type tissue) and not directly from the ovarian surface epithelium (mesothelium). Therefore, the ovary is involved secondarily³⁵.
Table 2. Clinicopathologic and molecular features of Type I and Type II epithelial ovarian carcinomas (Adapted from references 35–37).
| Type I | Type II | |
|---|---|---|
| Usual clinical behavior | Clinically indolent (slow growing) | Highly-aggressive |
| Tumor grade | Low-grade (except clear cell carcinoma considered high-grade) | High-grade |
| Stage | Low stage | Advanced-stage |
| Shared lineage between cystic neoplasms and the corresponding carcinomas through and intermediate (borderline tumor) step supporting the morphologic continuum of tumor progression | Subtle morphological differences. Considerable overlap in the diagnosis by different pathologists. Exhibit papillary, glandular and solid patterns and diagnosed depending on the dominant pattern | |
| Stepwise sequence borderline-carcinoma |
Type I
Low-grade serous
Low-grade endometrioid
Clear cell
Mucinous
Type II
High-grade serous
High-grade endometrioid
Undifferentiated carcinomas
Malignant mixed mesodermal tumor (carcinosarcomas)
25% — 75% epithelial ovarian cancers
Genetically stable — Genetically unstable
Main specific mutations
Type I:
KRAS, BRAF (low-grade serous and mucinous)
Wnt-signaling pathway (CTNNB1), PTEN, PIK3CA, ARID1A, KRAS (low-grade endometrioid)
ARID1A, PIK3CA, KRAS (clear cell)
Distinctive mutation pattern that occurs in specific-cell type
Type II:
p53 >80% high-grade serous
CCNE1
BRCA1, BRCA2
Greater morphological and molecular homogeneity
Precursor lesion
Type I:
Atypical proliferative (borderline) tumors
Endometriosis (Atypical?)
Type II:
Serous Tubal intraepithelial carcinoma (STIC)
Type I tumors represent only 25% of epithelial ovarian cancers and mostly arise from well-established benign (premalignant) precursors, namely from endometriosis or from atypical proliferative (borderline) tumors²⁸³⁵. This group includes the so-called endometriosis-related ovarian cancers, namely endometrioid and clear cell carcinomas³⁶.
1) RETROGRADE MENSTRUATION: THE MECHANISTIC THEORY FOR ENDOMETRIOSIS AND OVARIAN CARCINOGENESIS
The underlying mechanisms of endometriosis have yet to be determined. Despite numerous theories attempting to clarify their nature, none of them can fully explain the heterogeneity of the disease³⁸³⁹. Interestingly, regarding the endometriosis-related ovarian carcinogenesis the Sampson’s initial theory of retrograde menstruation⁴⁰ has gained special interest. According to this theory, exfoliated viable endometrial fragments and cells are driven through the fallopian tube reaching the peritoneal cavity and/or the ovarian surface. Actually, retrograde passage of cells through the fallopian tube has been also implicated in the current model for ovarian carcinogenesis³⁵. It is possible that retrograde passage of blood and cellular material probably contain epithelial cells of the cervix, endometrium, or of the fallopian tube to the ovarian surface or into the lumen of a recently ruptured follicle. Indeed, most of epithelial ovarian cancers mimic the cellular properties of the cervical, endometrial or tubal epithelium³⁵. Therefore, it is also possible that this retrograde travel carries cancer progenitor cells into the ovarian parenchyma⁴¹⁴². Of further interest has been the demonstration that eutopic endometrium in women with endometriosis exhibits intrinsic molecular abnormalities, such as mutations in several cancer-driver genes and activation of known oncogenic pathways⁴²⁴³.
Several studies have demonstrated a decrease in both endometriosis development and ovarian cancer risk after hysterectomy, tubal ligation or salpingectomy¹⁰⁴¹⁴⁴⁵ possibly attributable to the prevention of retrograde passage of carcinogenic factors from the uterus. In addition, animal models using genetically engineered mice, indicate that needle pass through the uterus/oviduct is required for the formation of peritoneal endometriosis or epithelial ovarian cancer⁴⁶. Such finding confirms the role of tubal patency in the genesis of endometriosis and associated ovarian cancer. Specifically, for endometriosis-associated histotypes, tubal ligation is associated with a 60% of risk reduction for endometrioid tumors (Relative Risk (RR) 0.40 [95% CI, 0.30–0.53])⁴⁷. The fact that tubal ligation, a procedure in which the fimbriae is preserved, results in a higher risk reduction for endometrioid carcinoma⁴⁷ strongly suggests that the precursor cell of EAOC come from the uterus (endometrium) and not from the tube.
The results of the Nurses’s Health Study⁴⁸ demonstrated a 76% increase in risk of ovarian cancer in intrauterine device (IUD) users respect to non-users (RR 1.76 [95% CI, 1.08–2.85]). The increase in risk was specifically associated with endometrioid (RR 2.40) and serous (RR 2.17) subtypes. This is very interesting since IUD users should be, in general, fertile and parous i.e. at reduced risk. This finding has been interpreted in terms of increased likelihood of tubal and peritoneal infection/inflammation, but it can also be associated with the increase in monthly blood loss in IUD users, which possibly leads to an increased transtubal retrograde menstrual flow⁴⁹.
Ovulation not followed by pregnancy is an accepted risk factor for ovarian cancer⁵⁰⁵¹. Despite ovulation is a physiological process, the “incessant ovulation theory” proposed for Fathalla in 1971, suggests that the repetitive damage and subsequent repair of the ovarian epithelium during ovulation may elevate the risk of cancer development. Therefore, the risk of ovarian cancer increases with the numbers of ovulations⁵⁰. Indirect epidemiological evidence supports this concept and multiple factors that alter the ovulation cycle, such as the use of oral contraceptive pills (OCP), are related to a reduced risk of ovarian cancer¹⁰⁴⁵. Specifically, for endometriosis-associated histotypes, OCP use is associated with a 27.1% and 21.3% of risk reduction for endometrioid and clear cell carcinomas, respectively⁵².
In summary, during ovulation, the ovarian surface epithelium is damaged during follicular rupture and subsequently repaired. During such repair invagination and inclusion cysts are formed in the cortical stroma, providing the opportunity for a stepwise sequence of genetic alteration⁵¹. In parallel, if endometrial tissue fragments line up along such inclusion cyst wall or became trapped on a recently ruptured follicular cyst, this arrangement could eventually evolve into an ovarian endometrioma, which over time, accumulates blood and inflammatory products. Later on, these chronic inflammatory processes could result in atypical endometriosis and/or an ovarian carcinoma (see below Inflammation).
Interestingly, an alternative origin of ovarian endometrioma has been hypothesized to be consequence of coelomic (Müllerian) metaplasia. According to this theory the coelomic epithelium (mesothelium) covering the cortical inclusion cysts could undergo a metaplastic change into endometrium³⁹. Inclusion cysts have also been postulated as the site origin of epithelial ovarian cancers.
Another possibility is the malignant transformation of endometriotic lesions localized in the tube. The presence of endometriosis in the tubes is a frequent reason for tubal obstruction, adhesions, hydrosalpinx development, and subfertility among endometriosis patients. The prevalence of tubal endometriosis ranged from 6.9 to 69%⁵³. Noteworthy, the coexistence of tubal endometriosis and endosalpingiosis (the presence of ectopic tubal epithelium) have been also identified to be a risk factor for EAOC⁵⁴.
2) GENETIC ALTERATIONS IN THE ENDOMETRIUM, ENDOMETRIOSIS AND ENDOMETRIOSIS-ASSOCIATED OVARIAN CANCER
As technology advanced, the capacity to perform genome-wide analyses and the opportunity for whole exome and RNA sequencing to assess somatic mutations have developed. Current evidence demonstrates that eutopic endometrium, benign endometriosis (non-cancer-associated) and EAOC carry distinct genetic alterations and pathways dysregulations that distinguish them from non-EAOC. These genes can be grouped into two categories: tumor suppressor genes and oncogenes.
Tumor suppressor genes encode for proteins involved in cell cycle regulation and apoptosis. When both copies of this genes are mutated, abnormal cells are able to replicate out of control, leading to cancer. Loss of heterozygosity (LoH) occurs when only one copy of the gene is lost. Thus, a single copy of the lost gene still remains. On the other hand, oncogenes are mutant gene forms that when activated contribute to cancer development via uncontrolled cellular growth and division.
2.1) CANCER-ASSOCIATED MUTATIONS IN EUTOPIC ENDOMETRIUM
The presence of somatic mutations in eutopic endometrium of women with and without endometriosis has been demonstrated by several studies. Such alterations include dysregulation in cancer-driver genes, including PTEN, KRAS, PIK3CA and abnormal expression of key components of cancer-related signaling pathways, such as PI3K/Akt/mTOR and Wnt/β-catenin pathways⁵⁵⁵⁷.
Lac et al⁵⁵ evaluated the presence of somatic driver mutations within histologically unremarkable eutopic endometrium. They found oncogenic mutations in over 50% of normal endometrial samples, including KRAS, PTEN, PIK3CA among others. However, no patients in this study exhibited loss of ARID1A gene, which is known to be involved in endometriosis and EAOC (see below).
Li et al⁵⁸ compared genetic profiles of epithelial cells from healthy endometrial epithelium, ovarian endometriotic epithelial cells, and matched eutopic endometrial epithelial cells. Most identified genetic variants were associated with cell adhesion, and chromatin remodeling and were present in endometriosis and matched eutopic endometrium. Moreover, ARID1A was altered in both eutopic endometrial and ovarian endometriotic epithelial cells. However, despite cancer-associated genes mutations are prevalent in normal endometrium, studies have suggested that they remain in a subclonal and mosaic-like state in which single endometrial glands harbor distinct mutation profiles⁵²⁵⁹. Additionally, the authors reported several additional genetic alterations predicted to alter protein structure in eutopic endometrial epithelial cells from disease-free women⁵⁸.
Although endometriotic tissue and normal eutopic endometrium are histologically similar and even could share the same oncogenic mutations, they are not identical: a distinct mutational profile and a different mutant allele frequency have been noted between them⁴². While normal endometrial epithelium usually carries one driver mutation per gland⁵⁹, this number is increased in endometriotic lesions⁴²⁶⁰. It has been shown that the development of cancer driver mutations in endometriotic lesion often occurs during the first years of life⁵⁹ and that the likelihood of a woman to have a somatic mutation increased by 5% per year⁵⁹. Despite the mutagenic burden progressively increases with age, it has been shown to decrease with parity⁵⁹.
2.2) CANCER-ASSOCIATED MUTATIONS IN ENDOMETRIOSIS (TYPICAL OR ATYPICAL) WITHOUT CANCER
Most studies of somatic mutations in endometriosis have been restricted to endometriosis with concurrent cancer⁶¹. These studies suggested that driver mutations shared by both lesions were the mutations responsible for the progression of the endometriosis to cancer⁶¹⁶². Nonetheless, only a few studies have examined the occurrence of somatic cancer driver mutations in patients with endometriosis without cancer⁴²⁶⁰⁶³⁶⁶.
In 2018, Zou et al⁶⁷ reported for first time the presence of classical cancer driver gene mutations in benign endometriosis. Moreover, in this study the co-occurrence of KRAS and ARID1A mutation was also identified for first time in a single individual. To date, the most commonly reported cancer-involved genes mutations found in benign endometriosis (typical/ atypical) are the tumor suppressor genes PTEN, ARID1A, the oncogene KRAS and the PPP2R1A gen, (which encodes a regulatory subunit of protein phosphatase 2, implicated in chromosome segregation and the negative control of cell cycle)⁶⁰⁶⁵⁶⁷.
Sato et al⁶⁴ investigated the potential role of PTEN gen in endometriosis-associated ovarian carcinogenesis in 34 patients with benign endometriomas. The authors reported a LoH in PTEN gen region and PTEN mutations in 56.5% and 20.6% of patients, respectively. Based on these finding the researchers suggested that inactivation of PTEN tumor suppressor gene is an early event in EAOC development.
A recent study using next-generation sequencing to determine whether benign, but invasive, non-ovarian deep endometriosis lesions harbor somatic mutations associated with cancer⁶⁰. Surprisingly, the majority of lesions harbored somatic mutations, and 26% harbored known cancer driver mutations, including ARID1A, PIK3CA, KRAS and PPP2R1A. Given the low rate of malignant transformation of endometriosis these results suggest that the presence of such mutations alone is neither sufficient to drive malignant transformation of endometriosis nor indicative of likely progression to cancer⁶⁰. Moreover, the fact that a minority of analyzed lesions harbored detectable mutations, as it has also been observed in patients with endometriosis co-occurring with cancer⁶¹, potentially supports the existence of multiple endometriosis lineages within the same patient. Another remarkable finding of this study was that all cancer driver mutation were present only in the epithelium and not the stroma in the same lesion, giving a potential role to epithelial compartment in the emergence of distinct clonal cell populations⁶⁰. Since ovarian cancer originates from epithelial cells, we think it is important to consider this distinction.
Not surprisingly, the described genetic alterations have been specifically reported among patients with atypical endometriosis⁶¹⁶⁹⁷⁰. A recent prospective study, using loss of BAF250a expression as a surrogate marker for ARID1A mutation, showed a significant higher loss of BAF250a expression in atypical endometriosis compared with typical endometriosis. Although loss of BAF250a expression in patients with architectural atypia was 40% vs. 9% in patients with cytologic atypia, the difference was no significant (p<0.149)⁶.
In the same line, Er et al⁷⁰ identified several novel genetic mutations in several pathways involving in tumorigenesis among patients with EAOC. Notably, in most of patients, identical somatic mutations were detected in atypical endometriosis and tumor lesions. Such alterations were also present in non-atypical endometriotic lesions in two patients indicating the presence of genetic alterations in preneoplastic lesions.
2.3) MAIN GENES AND SIGNALING PATHWAYS INVOLVED IN ENDOMETRIOSIS-ASSOCIATED OVARIAN CANCER BIOLOGY
Targeted next-generation sequencing has allowed the identification of genetic aberrations in endometriosis involving multiple pathways promoting malignant transformation and cancer cell growth⁷⁰. Such alterations involve the PI3K/Akt/mTOR, the chromatin remodeling, the Wnt, the ERK/MAPK and the Notch signaling pathways. Moreover, several genes involved in aberrant DNA repair mechanisms, cell cycle control, apoptosis and mismatch repair (MMR) systems have proven to be dysregulated in EAOC⁷⁰ (Fig 1). Notably, the existence distinct genetic profile between endometriosis-associated and non-endometriosis associated endometrioid ovarian cancers has been proposed⁷¹. In this review we will discuss only the most widely studied and the most relevant signaling pathways in the pathogenesis of EAOC.
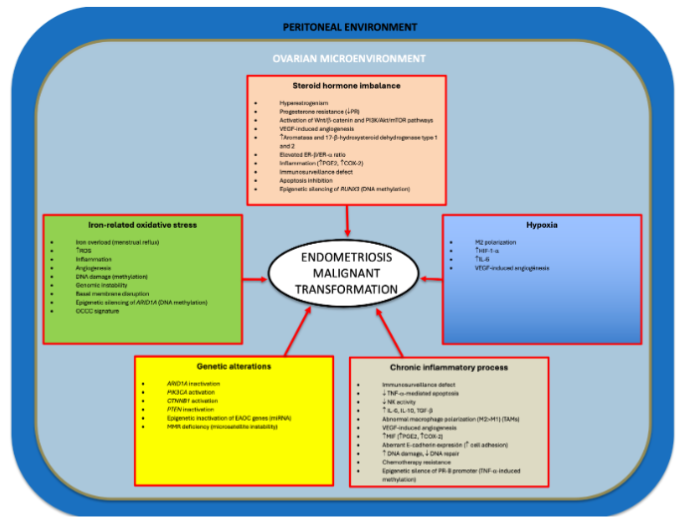 Fig 1. Mechanisms implicated in endometriosis malignant transformation.
Fig 1. Mechanisms implicated in endometriosis malignant transformation.Note that several processes are triggered by different mechanisms (crosstalk) and that epigenetic alterations can be induced by steroid hormones, chronic inflammation, and oxidative stress.
2.3.1) ARID1A
AT-rich interactive domain-containing protein 1A (ARID1A) gene is a tumor suppressor genes that encodes BAF250a protein, one of the subunits of SWI-SNF chromatin remodeling complex, which alters the accessibility of chromatin to different nuclear factors, thereby preventing genomic instability. ARID1A is mutated in a wide range of cancers, especially in those arising from ectopic and eutopic endometrium, including EAOC⁷²⁷³.
In fact, downregulation of ARID1A is thought to induce malignant transformation of endometriosis in a stepwise manner, as a gradual loss of ARID1A expression has been observed from benign (20%) to atypical endometriosis (40%) and to ovarian clear cell carcinoma (58%)⁷⁴.
Current evidence favors the role of ARID1A somatic mutation as a major molecular contributor to clear cell and endometrioid ovarian carcinomas⁶¹⁶⁷. Wiegand et al⁶¹ investigated ARID1A mutations and loss of BAF250a in EAOC. They found ARID1A mutations in 46% of ovarian clear cell carcinomas, 30% of endometrioid carcinomas and in none of high-grade serous carcinomas. In addition, loss of BAF250a was found in 73% of clear cell and 50% of endometrioid ovarian cancers with an ARID1A mutation compared to 11% of non-EAOC. Interestingly, two patients presented ARID1A mutations and loss of BAF250a expression in the tumor and in contiguous atypical endometriosis but not in distant endometriotic lesions. Based on these findings in preneoplastic lesions, several authors have suggested that both ARID1A disruption and loss of BAF250a are early events in endometriosis-associated ovarian carcinogenesis⁶¹⁶⁷⁷⁵.
Nevertheless, Yachida et al⁷⁷ reported that 16% of ovarian clear cell carcinomas and all benign endometriosis samples carrying ARID1A loss-of-function mutations preserved immunoreactivity for ARID1A. These data are consistent with the “two-hit” hypothesis, meaning that both alleles of the ARID1A gene must be inactivated in order to cause a phenotypic change.
Lakshminarasimhan et al⁷⁸ studied the effect of ARID1A loss in non-tumorigenic endometriotic cells. Interestingly, the authors reported the development of a tumoral-like phenotype characterized by an enhanced growth and invasion capacity, suggesting that ARID1A mutation could be a necessary step in the malignant transformation of endometriosis. Moreover, several genes presented altered expression upon downregulation of ARID1A and most of such expression changes were accompanied by alterations in histone modifications (see epigenetics). Hence, loss of ARID1A in endometriotic cells is sufficient for the induction of epigenetic, molecular and phenotypic alterations indicative of potentially oncogenic transformation.
Interestingly, loss of ARID1A expression is reported in 33% of ovarian seromucinous carcinomas (OSMC) (mixed Müllerian carcinoma)⁷⁹, which is similar to their frequency the other EAOC (clear-cell and endometrioid), providing compelling evidence to include them in the group of endometriosis-related neoplasms³⁶⁷⁹. However, the 5th edition of World Health Organization (WHO) Classification of Female Genital Tumors published in 2020 has removed OSMC as a distinct entity and now considers it as a subtype of ovarian endometrioid carcinoma⁸⁰. Such modification generated some controversy, and recent research argues that the different clinical features and prognosis of OSMC make it not suitable to be directly classified as ovarian endometrioid carcinoma⁸¹.
Nonetheless, ARID1A mutations seem not sufficient on their own to cause cancer⁸²⁸⁴. ARID1A mutations frequently co-occur with mutations leading to an activation of PI3K/AKT pathway (see below), such as mutations in PIK3CA, suggesting a cooperating mechanisms (crosstalk) between these two pathways in endometriosis-associated ovarian carcinogenesis⁷²⁷³⁸⁶ (Fig 2).
Further evidence demonstrating a crosstalk between pathways comes from genetically engineered mouse models that revealed that the sole loss of ARID1A gene function does not induce ovarian cancer⁸⁷, but when the ARID1A and PTEN genes are simultaneously knocked out, 59% of mice develop ovarian endometrioid or undifferentiated carcinoma, and 41% exhibit hyperplasia of ovarian surface epithelium⁸⁴. On the other hand, co-mutation of ARID1A and PIK3CA leads to formation of clear cell-like ovarian tumors through sustained interleukin (IL)-6 overproduction, suggesting a protective effect of ARID1A against inflammation-driven tumorigenesis⁸⁷ (Fig 2).
Despite ARID1A mutation have been associated with an unfavorable prognosis in several malignancies⁸³⁸⁸, loss of ARID1A has not been identified as prognostic factor for ovarian cancer⁸³⁸⁹.
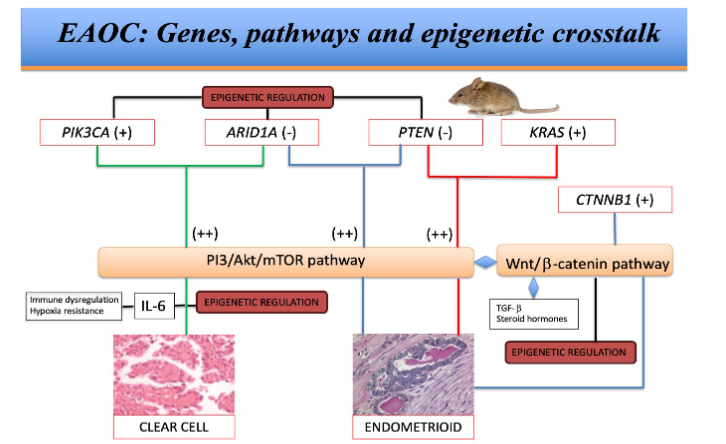
Fig 2. EAOC: Genes, pathways and epigenetic crosstalk
Genetic, metabolic and epigenetic interactions in EAOC. Note that cooperation between KRAS mutation and PTEN inactivation has not been described in humans.
(+) Activation
(++) Cooperating mechanism
(-) Inactivation
◆ Crosstalk
2.3.2) PIK3CA
PIK3CA is a gene encoding for the α-subunit of phosphatidylinositol-3 kinase (PI3K). Active mutations in PIK3CA have shown to upregulate the PI3K/Akt pathway signaling. Previous studies have demonstrated that this gene is dysregulated in up to 45% of clear cell and endometrioid ovarian cancers. While PIK3CA mutations have been identified in 20% of clear cell and 20% of endometrioid ovarian cancers compared to only 2.3% of serous carcinomas (p<0.001)⁹¹.
Yamamoto et al⁹² demonstrated the presence of identical PIK3CA somatic mutations in 43% of clear cell carcinomas and in 90% of cases of coexisting endometriosis adjacent to carcinomas. Moreover, in most of cases, such mutations were identified in endometriotic tissue without evidence of atypia.
Based on the finding of PIK3CA mutations in non-atypical precursor lesions, the authors concluded that mutation in the PIK3CA gene is early event in the malignant transformation of endometriosis.
Kuo et al⁹³ reported PIK3CA activating mutations in 33% of ovarian clear cell carcinomas, but the percentage of such mutations reached 46% when affinity purified clear cell tumors and clear cell carcinoma cell lines are included. In the same line Huang et al⁸⁵ identified PIK3CA mutations in 34% of ovarian clear cell carcinoma. On the other hand, a recent review reported PIK3CA mutation in 40% of endometrioid ovarian carcinomas⁸⁶.
Taken together, these findings give direct evidence that PIK3CA is an oncogene in EAOC.
2.3.3) PTEN AND KRAS
Phosphatase and tensin homolog (PTEN) acts as a tumor suppressor gene through the action of its phosphatase protein product, involved in the regulation of the cell cycle. PTEN is the natural inhibitor of the PI3K/AKT signaling pathway. Inactivating mutations of PTEN are reported in 15 to 20% of endometrioid carcinomas and in almost 10% of clear cell carcinomas³⁶.
An earlier study by Dinulescu et al⁴⁶ described an interesting mice model of peritoneal endometriosis and endometrioid ovarian carcinoma, based on the activation of the oncogene KRAS and conditional PTEN deletion. While the expression of KRAS or conditional PTEN deletion in the ovarian surface epithelium gave rise to preneoplastic ovarian lesions with endometrioid glandular morphology, the combination of the two mutations leaded to the rapid development of metastatic ovarian carcinomas. Despite KRAS mutations can be found in approximately 10% of EAOC, recent evidence suggests that they are more related to the development of endometriotic lesions⁹⁴. Additionally, KRAS mutations are more prevalent in non-endometriosis-associated type I ovarian cancers, especially mucinous carcinomas³⁶. Based on this finding, it has been proposed that KRAS mutations may be associated with late events in the malignant transformation of endometriosis⁹⁴.
In addition, data supporting cooperation between KRAS mutation and PTEN inactivation in human endometrioid ovarian carcinomas are lacking⁹⁵. Since PTEN is under-expressed in most endometriosis samples⁹⁶, regardless of the occurrence of cancer, it seems that whereas inactivating mutation of PTEN is not sufficient to induce malignant transformation of endometriotic cells on its own, it opens the way for subsequent genetic events that may lead to carcinogenesis.
2.3.4) PI3K/Akt/mTOR PATHWAY
The phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt)/mammalian target of rapamycin (mTOR) pathway is an intracellular signaling axis important in regulating the cell cycle. It is extremely multipart owing to the alterations within the pathway itself and in their inputs⁹⁶. This pathway and their downstream components are directly related to cellular quiescence, proliferation and longevity, and it is considered one of the main pathways in ovarian cancer⁹⁶⁹⁷. Amplifications or mutations in PI3K/Akt/mTOR pathway is involved in several cancers. However, this pathway is also deeply involved in the normal cyclical endometrial function and has been implied in the pathogenesis of endometriosis⁹⁸. As mentioned before, inactivating mutations in PTEN, ARID1A and activating mutations of PIK3CA genes, frequently found in EAOC, can lead to abnormal and synergistic activation of the PI3K/AKT pathway⁷²⁷⁵⁸⁶.
In summary, similar genetic alterations PI3K/Akt/mTOR pathway components occur in eutopic normal endometrium, endometriosis and EAOC. However, the role of such defects in the progression of endometriosis to ovarian cancer remains unclear. It has been hypothesized that spontaneous and repeat PI3K/Akt/mTOR pathway-associated mutations arising through normal endometrial glands may act as precursor events that, upon the emergence of additional genetic abnormalities can lead to EAOC, via the intermediary stage of endometriosis⁸.
2.3.5) CTNNB1 GENE AND Wnt/β-CATENIN PATHWAY
Another gene involved in endometriosis-associated ovarian carcinogenesis is the oncogene CTNNB1 which encodes β-catenin, a dual function protein involved in regulation of cell-cell adhesion (as a subunit of cadherin protein complex) and gene transcription. It also acts as an intracellular signal transducer in the wingless-type integration site (Wnt) signaling pathway. This complex pathway is involved in several cell functions, such as cell proliferation and differentiation, apoptosis, cell migration, and stem cell maintenance in adults, therefore it has been implicated in cancer initiation and progression. Moreover, Wnt/β-catenin pathway is involved in ovarian embryogenesis, is thought to be involved in epithelial-mesenchymal transition (EMT). A detailed description of this pathway is beyond the scope of this review and can be found elsewhere⁹⁸.
In the gynecologic field, Wnt/β-catenin pathway plays a pivotal role in endometrial physiology maintaining the monthly balance between estrogen-dependent proliferation and progesterone-induced differentiation⁹⁹. In the same line Wnt/β-catenin pathway dysregulations have been also implicated in both the pathogenesis and invasion capacity of endometriosis¹⁰⁰¹⁰¹. Alterations in the Wnt/β-catenin pathway have shown to play important roles in the tumorigenesis of ovarian cancer⁹⁸.
Endometrioid ovarian carcinomas often harbor activating mutations in the CTNNB1 gene (16–54%). Despite CTNNB1 gene mutations are rare in serous and clear cell carcinomas⁹⁸, nuclear β-catenin has been observed in serous and clear cell ovarian cancers⁹⁸. CTNNB1 mutations are associated with squamous differentiation, low tumor grade and favorable outcome in endometrioid ovarian cancers³⁶⁹⁵.
It is noteworthy that Wnt/β-catenin pathway has several crosstalk with other pathways involved in both endometriosis¹⁰¹ and ovarian cancer⁹⁵, such as PI3K/Akt/mTOR, transforming growth factor β (TGF-β) and steroid hormone signaling pathways. Further, the epigenetic influences on the Wnt/β-catenin pathway are well documented¹⁰².
Wu et al⁹⁵ demonstrated that activation of both Wnt/β-catenin and PI3K/Akt/mTOR pathways (via PTEN inactivation or PIK3CA activation) often cooperate in endometrioid ovarian cancer pathogenesis in humans, and that 66% of endometrioid ovarian carcinomas with PTEN mutations also have Wnt/β-catenin pathway defects. These tumors were typically low-grade, had occasional foci of squamous (epithelial) differentiation (positives for cytokeratin 8 and 19), and significant areas of less-differentiated mesenchymal-appearing cell (negative for cytokeratins, loss of E-cadherin immunoreactivity) suggesting of EMT. Notably, Wnt/β-catenin and PI3K/Akt/mTOR pathways defects have both been implicated in EMT, a process involved in both endometriosis and ovarian cancer¹⁰³¹⁰⁴.
Intriguingly, the widely accepted hypothesis that somatic mutations in cancer driver genes may induce the malignant transformation of endometriosis, has been recently challenged. Linder et al¹⁰⁵ explored the mutation and copy number profiles of endometriosis with a confirmed subsequent malignant association in 11 patients who developed either endometrioid or clear cell ovarian cancer later in life after their diagnosis of benign endometriosis. Surprisingly, no shared cancer-associated mutations were observed. Moreover, the most frequently found genetic alterations between benign endometrioma and paired EAOC were in genes related to inflammatory response and immunotolerance, suggesting that adaptation to inflammation is an early and crucial event in the development of EAOC.
3) PERITONEAL ENVIRONMENT, INFLAMMATION AND IMMUNOLOGICAL ASPECTS LINKING ENDOMETRIOSIS AND OVARIAN CANCER
Common components of endometriosis and ovarian cancer are the inflammatory pattern and immune system mobilization. Therefore, current molecular studies have aimed to establish links between endometriosis and EAOC through pathways related to inflammation, oxidative stress and hyperestrogenism.
It is well accepted that menstruation and ovulation represent physiological injury that triggers repetitive inflammatory reaction of the uterus, ovaries and peritoneum, thereby, demanding tissue repair and remodeling. Moreover, the correlation between chronic inflammation and cancer development is very known¹⁰⁶.
In healthy females, once endometrial cells reach peritoneal cavity during retrograde menstruation, they undergo apoptosis and are efficiently cleared by macrophages and natural killer (NK) cells—a phenomenon known as immune surveillance¹⁰⁶. In a parallel manner, in the ovarian surface epithelium, macrophages contribute to tissue repair and proliferation through the secretion of several growth factors, TGF-β and IL-10, as well as apoptosis via the secretion of Reactive Oxygen Species (ROS)¹⁰⁶.
In normal conditions, the presence of endometrial fragments in the peritoneum activates macrophages leading the onset of the inflammatory response. Macrophages present in different tissues are polarized according to changes in their environment, forming different macrophage subtypes, namely M1 and M2 macrophages¹⁰⁷. Both subtypes are closely related to inflammatory response. In general, M1 macrophages are mainly involved in pro-inflammatory responses producing inflammatory factors, such as IL-6 and tumor necrosis factor-α (TNF-α). On the other hand, M2 subtype are involved in tissue-repair, fibrosis and angiogenesis by the production of anti-inflammatory factors, such as IL-10 and TGF-β¹⁰⁶¹⁰⁸. However, studies have shown that there is a significant overlap in these profiles according to the tissue microenvironment and type of signaling¹⁰⁸.
In the context of endometriosis, cell-mediated immune responses can be disrupted, impairing this natural clearance process. Hence, endometrial cells can adhere to peritoneal surface, proliferate and activate an inflammatory response¹⁰⁶. The precise mechanism responsible for immunosurveillance evasion is not known, but it seems to be related to a dysregulation in apoptotic pathways. In addition, recent studies have shown that peritoneal fluid from patients with endometriosis increases proliferation, induces inflammatory genes, and modulates epigenetic pathways that may promote EAOC¹⁰⁹.
An alteration in tumor necrosis factor (TNF)-α-mediated apoptosis in ectopic endometrium has been documented in patients with endometriosis, and current evidence supports that this adaptative mechanism for apoptosis evasion is estrogen-dependent via stimulation of estrogen receptor (ER)-β¹¹⁰. TNF-α acts as a regulator of the pro-inflammatory microenvironment of ovarian cancer through the modulation of several cytokines that may promote ovarian tumorigenesis¹¹⁰. Additionally, TNF-α is differentially regulated in ovarian cancer cells and it has been found to have an increased expression in clear cell ovarian carcinoma compared to normal ovarian tissue¹¹⁰.
Another well documented immunologic alteration in patients with endometriosis is the reduced cytotoxicity of peritoneal NK cells¹⁰⁶. This NK cell dysfunction results from a complex interplay of cytokines within the intricate microenvironment of endometriotic lesions. Yang et al¹¹¹ proposed that the interactions between macrophages and endometrial stromal cells could downregulate NK cell cytotoxicity through the secretion of IL-10 and TGF-β by the interacting cells. Additionally, increased levels of IL-6 in peritoneal fluid of patients with endometriosis might contribute to the suppressed NK cell activity¹⁰⁶¹¹².
Macrophages is the one of the main cell population in invasive tumors and they seem to be “educated” by tumors cells to promote tumor immune escape, angiogenesis, tumor growth, and metastasis. It is believed that macrophage polarization in tumors is driven by clues in the tumor microenvironment, such as low pH, inflammatory and cytokine profile, hypoxia and extracellular matrix characteristics¹⁰⁷. Notably, these so-called tumor-associated macrophages (TAMs) have the function of killing several population cells within the tumor, such as fibroblasts and other macrophages, whereas promoting tumoral cell growth and pro-tumoral conditions. M2 macrophages are similar in phenotype to TAMs expressing metalloproteinases and other growth factors such as epithelial growth factor (EGF), vascular endothelial growth factor (VEGF) and TGF-β. Such molecular profile facilitates cancer cell proliferation, invasiveness, epithelial-mesenchymal transition and metastasis formation¹⁰⁷.
Endometriosis is characterized by a macrophage polarization into M2 phenotype, which seems to be beneficial to endometriosis development. For example, secretion of VEGF favors angiogenesis, and the secretion of anti-inflammatory cytokines, such as IL-10 and TGF-β, contributes to the growth of endometriosis lesions by impairing the cytotoxicity of NK cells.
Published data demonstrate that TGF-β has a pivotal role in myofibroblastic differentiation, fibronectin and collagen synthesis, and fibrosis promotion, distinct features of endometriosis progression¹⁰⁶.
113,114. Intriguingly, TGF-β has been also involved in the development of ovarian cancer¹¹⁵,¹¹⁶.
Interestingly, TGF-β has been implicated in the development of peritoneal endometriosis¹¹⁷,¹¹⁸, endometriosis invasion capacity¹¹⁹, and ovarian surface epithelium neoplastic transformation¹¹⁵.
Other relevant molecule is the macrophage migration inhibitory factor (MIF). MIF is found in elevated concentrations in peritoneal fluid of women with endometriosis and active endometriotic lesions. MIF has a direct role in the upregulation of COX-2 synthesis and prostaglandin E2 (PGE2) secretion via p38 kinase activation in ectopic endometrial cells¹²⁰. Thus, inducing a proinflammatory phenotype in endometriotic tissues.
In both endometriosis and ovarian cancer, the cells have an abnormal adhesion capacity, which allows them to develop distant endometriotic lesions and metastasis, respectively. Cell adhesion is mediated by several molecules, such as TGF-β, integrins and cadherins¹²¹. Endometriosis is reported to have a highly variable and aberrant integrin expression compared with eutopic endometrium. These alterations include molecular mechanisms of invasion and metastasis shared with carcinoma cells, such as an aberrant level of E-cadherin expression¹²². As mentioned before, β-catenin is a subunit of E-cadherin highly expressed in ovarian cancer and endometriosis⁹⁸.
In summary, the chronic and aberrant expression of cytokines alters several regulatory signaling pathways, which facilitate both endometriosis progression and cancer growth, invasion and metastasis through DNA damage and inhibition of DNA repair (Fig 1). Hence, resulting in accumulation of genetic mutations in endometriotic cells that may favor malignant transformation.
4) HYPOXIA AND INTERLEUKIN-6 CONTRIBUTION AS A CARCINOGENIC DRIVER IN ENDOMETRIOSIS
Due to the rapid tumoral growth, hypoxia is an intrinsic feature of tumor microenvironment. Hypoxia is involved in the induction of epithelial-metabolism, TAM infiltration and have a profound effect on macrophages polarization into M2 profile. Interestingly, M2 phenotype TAMs have been involved in the survival adaptation of tumor cells preparing them for an impending hypoxic injury before changes in oxygen availability. Such adaptation is attributed to an increased IL-6 production by M2 TAMs¹²³. Additionally, IL-6 facilitates tumor cell survival, induce cancer stemness, prime M1 macrophages towards M2 phenotype and triggers the expression of key angiogenic factors, such as hypoxia-inducible factor (HIF)-1α, and VEGF¹⁰⁶ (Fig 1). Notably, VEGF-driven angiogenesis is the target of some immunotherapy drugs currently use in the treatment of ovarian cancer¹²⁴.
As mentioned before, IL-6 overproduction was also involved in the pathogenesis of clear cell ovarian carcinoma. Chandler et al⁸⁷ reported that synchronous ARID1A loss and PIK3CA overactivation synergistically induced upregulation of IL-6. IL-6 secretion triggers and it triggered by the JAK/STAT pathway, thereby initiating a signaling cycle that promotes tumor cell growth and differentiation¹²⁵. Furthermore, IL-6 is the direct target of ARID1A tumor-suppressor activity and under the absence of the negative regulation of the latter, coexisting amplification of PIK3CA promotes IL-6 overexpression, thus maintaining the JAK/STAT signaling loop⁸⁶. Therefore, the synergistic action of ARID1A loss and PI3K/Akt/mTOR pathway upregulation in the malignant progression of endometriosis can be partially explained by their cooperation in activating IL-6 and thus in promoting a pro-tumorigenic inflammatory cytokine signaling. Consequently, IL-6 is highly expressed in ovarian cancer and its levels have been correlated to tumor angiogenesis, cancer progression and chemotherapy resistance¹²⁵.
In the same line, the evaluation of the tissue immune microenvironment has revealed a specific role for complement proteins in the malignant transformation of endometriotic tissue. Suryawanshi et al¹²⁶ demonstrated that concomitant KRAS activation and PTEN deletion leaded to upregulation of complement proteins in epithelial cells. They also found that different immune “profiles” exist for each eutopic endometrium, endometriosis and EAOC. Because of the sensitivity and specificity of such “profile” in detecting early molecular changes in cells undergoing malignant transformation, it is possible that immune system profiling could lead to early detection of women at risk of EAOC.
5) IRON-RELATED OXIDATIVE STRESS AND MALIGNANT TRANSFORMATION OF ENDOMETRIOSIS
Oxidative stress occurs when the production of ROS exceeds the capacity of cellular antioxidant defenses to remove these toxic agents. An increasing body of evidence suggests that oxidative stress within endometriosis is likely to contribute to the malignant transformation process by a fine-tuning balance between pro-oxidant iron overload (inflammatory) and antioxidant defenses (anti-inflammatory)¹²⁷.
Iron accumulation in the peritoneal cavity, stemming from retrograde menstruation, holds a particular interplay with macrophages activation and plays a role in the pathogenesis of endometriosis. Macrophages in the pelvic cavity carry out erythrocyte phagocytosis and iron metabolism, thereby resulting in elevated iron concentrations within the peritoneal fluid.
In patients with endometriosis, it is not known if these peritoneal protective mechanisms are originally defective or if they become overwhelmed by the amounts of hemoglobin in the liquid. As a result, iron overload can trigger oxidative stress, ROS generation and contribute to chronic inflammation and angiogenesis, thus leading to increased proliferative capacity of endometriotic lesions. Prolonged exposure to oxidative stress and high ROS levels may induce DNA damage (methylation) and genomic instability, facilitating the development of mutations that could induce the ovarian carcinogenesis process¹²⁸. Additionally, iron-related oxidative stress contributes to the peritoneal mesothelium and peritoneal basal membrane disruption, exposing sub-peritoneal collagen matrix, favoring the adhesion and metastasis of endometriotic and tumoral cells¹²⁶,¹²⁸ (Fig 1).
Notably, Yamaguchi et al¹²⁹ reported significantly higher free iron concentrations and increased oxidative DNA damage in the epithelial and stromal cells of endometriotic cysts when compared to non-endometriotic cysts and normal serum, proposing such iron overload as a possible cause of carcinogenesis in the endometriomas through the iron-induced persistent oxidative stress.
The exact mechanism why ovarian, but not deep or peritoneal endometriotic lesions, is the site of origin of most endometrioid and clear cell carcinomas is not known, but it seems to be related with the prolonged exposure to blood and the peculiar characteristics of ovarian microenvironment in favoring the initiation of genetic alterations⁴¹,¹³⁰ (Fig 1). The plasticity of ovarian surface epithelium, regarding continuous repair of ovulation wounds, could contribute to the susceptibility of these cells to oxidative stress from blood product degradation.
Hence, in case of EAOC, the real carcinogenic factor in clear cell and endometrioid cancers could be iron⁴¹.
Further evidence supporting the role of oxidative stress in the development of clear cell carcinomas come from the identification of a specific gene expression for these tumors. The so-called clear cell signature included several genetic alterations in genes involved in oxidative stress and inflammation, indicating that clear cell carcinoma specifically expresses stress-responsive genes¹³¹. Moreover, such gene signature was induced upon iron exposure in immortalized ovarian surface epithelial cells, supporting the transition from precursor cells to cancer¹³¹.
In summary, endometriosis is vulnerable to oxidative stress inducing DNA damage, apoptosis and cell death. On the other hand, iron overload induces signaling of several antioxidant detoxification systems which protect endometriotic tissues from iron-mediated oxidative damage. Hence, while most endometriotic cells die under excessive oxidative stress conditions a small subset of cells will be able to survive by cellular antioxidants mechanisms. Therefore, the enhanced antioxidation capability or the exposure of cells to sublethal levels of ROS result in an adaptive, cytoprotective modulation of several survival signaling pathways. Such mechanisms can induce malignant transformation of endometriosis allowing the survival of cells with damaged DNA, promoting carcinogenesis¹²⁷,¹³². In the same line, it has been demonstrated that cancer cells have less oxidative stress than endometriotic cells, and that they express higher levels of antioxidant proteins, which detoxify ROS, facilitating the remodeling of tumor microenvironment and cancer progression¹²⁷.
So, transtubal reflux of endometrial cells would give origin to endometriosis, but the eventual malignant transformation of endometriosis would be caused by cyclical bleeding in the pelvis, and free iron inside pseudocysts filled with “old blood”⁴¹.
6) ROLE OF STEROID HORMONE IMBALANCE IN MALIGNANT TRANSFORMATION OF ENDOMETRIOSIS
Endocrine dysfunction, with estrogen dependency and progesterone resistance is a key feature of endometriosis. Estrogen is a known driver of endometriosis, contributing to both proliferation of the disease and inflammation. Conversely, resistance to progesterone impedes the ability of progestins to mitigate the progression of endometriotic lesions. Hyperestrogenism is a known risk factor for the development of ovarian cancer from endometriosis¹³³.
Estrogens elicit its physiological effect on target cells by binding to estrogen receptors (ERs). ERs classified into nuclear (classical) and membrane-bound ERs (mERs). Nuclear ERs are subdivided in ER-α and ER-β, encoded by different genes, ES1 and ES2, respectively. However, ERα is expressed primarily in the uterus, whereas ERβ is expressed primarily in the ovary. On the other hand, mERs are located in the nuclear membrane and act through a genomic-independent (non-classical) signaling thanks to the G-protein-coupled estrogen receptor (GPER)¹³⁴.
Nuclear (genomic) estrogen signaling has several interconnections with other pathways involved in endometriosis progression and ovarian cancer, such as Wnt/β-catenin signaling pathway¹³⁵. On the other hand, non-classical estrogen signaling activates several pathways involved in the pathogenesis of EAOC, such as the PI3K/Akt/mTOR pathway¹³⁰ (Fig 1).
It has been speculated that inactivation of ER-α and its target genes/pathways, the decreased progesterone receptor (PR) levels and the increased ER-β pathway may be the driving factors for endometriosis malignant transformation¹³⁶. Despite ERα signaling pathway is largely deactivated in EAOC, there is a subset of ERα-regulated genes that remain highly expressed¹³⁶. These genes are involved in cell proliferation and angiogenesis. Zhang et al¹³⁵ demonstrated that the VEGF expression in endometriotic cells was mediated by ERα through the Wnt/β-catenin signaling pathway. Hence, it has been hypothesized that they may contribute to estrogen-dependent transformation of endometriosis to EAOC.
Additionally, endometriotic stromal cells contain numerous epigenetic defects that favor estrogen overproduction, decreased estrogen degradation, progesterone resistance and endometriosis progression¹³⁷–¹³⁹ (See epigenetics and Table 2).
Moreover, several enzymes such as aromatase and the 17-β-hydroxysteroid dehydrogenase type 1 and 2, that favor local estrogen production and estrogen signaling, are differentially expressed in endometriosis compared with eutopic endometrium¹³⁷¹³⁹.
In response to estrogen signaling, endometriotic tissue undergoes a noteworthy upregulation in ER-β. This elevated ER-β/ER-α ratio within endometriotic stromal cells is linked to the downregulation of progesterone receptors and an upsurge in COX-2 and PGE2 levels, thereby playing a role in the development of progesterone resistance and inflammation characteristic of endometriosis. Besides inflammation, PGE2 has been shown to be involved in immune surveillance evasion and regulation of vital processes related to tumor growth, including angiogenesis and apoptosis inhibition¹⁰⁶.
The aforementioned alterations may contribute to the carcinogenic process in neighboring epithelial cells¹⁴⁰.
Guo et al¹⁴¹ demonstrated the role of epigenetic inactivation of the tumor suppressor gene RUNX3, by an estrogen-dependent promoter hypermethylation, as an early event in the malignant transformation of ovarian endometriosis. Interestingly, this epigenetic change was positively associated with the expression of ERα.
Progesterone generally represses ERs, inhibiting the effects of estrogens at the cellular level. Progesterone elicits its physiological effects on the target cells by binding to nuclear progesterone receptor (PR) isoforms, A (PR-A) and B (PR-B). The progesterone resistance of endometriosis can be related to the absence of the stimulatory PR-B and the presence of the inhibitory PR-A¹³⁸. Interestingly, several epigenetic alterations involving the PR gene (PGR), the downregulation of progesterone-responsive gene expression, and their related signaling pathways have been noted in endometriosis¹³⁹.
7) EPIGENETIC MODULATION IN ENDOMETRIOSIS-ASSOCIATED OVARIAN CARCINOGENESIS
Epigenetics is defined as all heritable changes in gene expression (phenotype) that are not coded in the DNA sequence. Unlike genetic changes, epigenetic changes are reversible, arise in a gradual manner, and do not change the DNA sequence, but they can change how the DNA sequence is “read” in a specific cell type. Epigenetic changes affect gene expression by silencing (switching off), switching on, and stabilizing genes. It is now established that epigenetic modifications play definite roles both in endometriosis development¹³⁷,¹⁴²,¹⁴³ and ovarian carcinogenesis¹⁴⁴,¹⁴⁵. Main epigenetic mechanisms involved in endometriosis-associated ovarian carcinogenesis are DNA methylation/demethylation, histone modifications, and non-coding microRNAs¹⁴⁶.
7.1) DNA methylation is the best understood and most extensively studied epigenetic alteration, and it is mediated by a family of enzymes known as DNA methyltransferases (DNMTs) that catalyze the transfer of a methyl group to DNA. Typically, hypermethylation in the promoter region of a tumor suppressor gene lead to gene inactivation. By contrast, hypomethylation of an oncogene promoter region can lead to abnormally high gene expression, thus promoting tumorigenesis. The exact mechanisms of demethylation of genomic DNA are poorly understood.
Current research has demonstrated the presence of aberrant methylation in the promoter region of several genes involved in the malignant transformation of ovarian endometriosis¹⁴⁴ (Table 3). In a recent study, Wang et al¹⁴⁷ demonstrated that estrogens up-regulates DNMT1 and leads to hypermethylation of RUNX3 in the malignant transformation of endometriosis. In the same line, it has been demonstrated that TNF-α induces hypermethylation of PR-B promoter in endometriotic cells, leading to lower expression level of PR-B and progesterone resistance¹⁴⁸. Additionally, ARID1A gene promoter hypermethylation has been reported in endometriosis, probably via ROS-upregulated DNMT1 gene expression¹⁴⁹. Hence, it is noteworthy that different stimuli such as steroid hormones, chronic inflammation, and oxidative stress can induce epigenetic modifications leading EAOC (Fig 1).
Notably, a growing body of evidence suggests a direct regulation of the genes encoding ERα, ERβ and PR by DNA methylation, leading an aberrant gene expression of these genes in endometriosis¹³⁹. Interestingly, current research has demonstrated that the aforementioned epigenetic changes related to malignant transformation of endometriosis, commonly expressed in EAOC patients, are also expressed in the eutopic endometrium of these women¹⁴². These data indicate that the risk of malignant transformation of endometriosis is more related to the biological characteristics of eutopic endometrium of endometriosis than that with the occurrence of endometriosis itself.
Table 3. Main aberrantly methylated genes involved in endometriosis malignant transformation (summarized from reference 144)
| Gene | Function | Epigenetic modification |
|---|---|---|
| Runt-related transcription factor 3 (RUNX3) | Gene regulation in cell proliferation and differentiation. Downregulated in endometriosis and EAOC. | Hypermethylated |
| MutL protein homolog 1 (MLH1) | Member of DNA mismatch repair (MMR) system. MMR system repairs the errors that normally occur during replication of repetitive DNA sequences. MMR deficiency causes microsatellite instability (MSI). Downregulated in endometriosis and EAOC | Hypermethylated |
| E-cadherin (CDH1) | Cell-cell adhesion, polarization and differentiation of epithelial tissues. E-cadherin acts as a tumor suppressor. Downregulated in endometriosis and EAOC. | Hypermethylated |
| AT-rich interactive domain-containing protein 1A (ARID1A) | Tumor suppressor gene involved in chromatin remodeling. Downregulated in endometriosis and EAOC. | Hypermethylated |
| Progesterone receptor-B (PGR) | Induces the differentiation of endometrial stromal cells to decidualized cells and epithelial glandular cells to the secretory phenotype. Downregulated in endometriosis favoring progesterone resistance. | Hypermethylated |
| Aromatase P450 (CYP19A1) | Catalyzes the conversion of androgens to estrogens. Upregulated in endometriosis. | Hypomethylated |
| Estrogen receptor-β (ESR2) | Endometriosis growth and ovarian tumorigenesis. Upregulated in endometriosis | Hypomethylated |
| Estrogen receptor-α (ESR1) | Downregulated in endometriosis and EAOC | Hypermethylated |
7.2) Histone modification
Histone modification is the second most important epigenetic factor that has a critical role in the regulation of gene expression. Histones are proteins that act as spools around which DNA winds to create structural units called nucleosomes. Histones prevent DNA from becoming tangled and protect it from damage. Histones can be modified in many ways in their N-terminal tail, including acetylation, phosphorylation, and methylation, among others. Histone methylation can determine either activation or repression of gene transcription; instead, histone acetylation determines gene activation.
Histone modifications have been involved in endometriosis pathogenesis, and endometriosis-associated ERα and PR downregulation¹³⁹. Additionally, they have also been identified to play a role in several oncogenic pathways. For example, histone modifications have shown to have a pivotal role in the development of clear cell carcinoma phenotype in ARID1A silenced endometriotic cell lines⁷⁸. Despite these findings, the role of this epigenetic mechanism in endometriosis-associated ovarian carcinogenesis is still ambiguous¹⁴⁴,¹⁴⁵.
7.3) MicroRNA (miRNA)
MicroRNA (miRNA) are a novel class of small nonprotein coding, single-stranded RNAs that regulates a high numbers of biological processes and their related pathways, such as post-transcriptional gene expression, via gene silencing with either translational repression or degradation of mRNA. Additionally, miRNAs can upregulate target genes by directly binding to their promoter. Several miRNAs have been directly associated with dysregulated gene expression in both endometriosis and ovarian cancer.
In a recent comprehensive review, by Gaia-Oltea et al¹⁵⁰ the authors provide updated information about miRNA families and their predicted target genes involved in EAOC. Notably, several miRNA families targeting genes involved in endometriosis-associated ovarian carcinogenesis, such as ARID1A, PTEN, CTNNB1 and PIK3CA, are commonly dysregulated in non-atypical and atypical endometriosis and EAOC. Furthermore, some of the dysregulated miRNAs have more than one of these genes in their target profile. For example, miR-221-3p that targets PTEN, CTNNB1 and ARID1A¹⁵⁰. Therefore, a single miRNA dysregulation can be responsible of simultaneous mutations that favor malignant transformation of endometriosis.
8) CLONAL SELECTION IN EUTOPIC ENDOMETRIUM IN THE PATHOGENESIS OF ENDOMETRIOSIS AND THE DEVELOPMENT OF ENDOMETRIOSIS-ASSOCIATED OVARIAN CANCER
Nowadays it is widely accepted that normal human endometrial glands and endometriotic lesions are composed by clonal cell populations, thus, each gland are derived from a common progenitor cell⁴²,¹⁵¹. Since most neoplasms are monoclonal in origin, monoclonality of endometriotic lesions and endometriomas suggests their neoplastic potential¹⁵²,¹⁵³.
Clonal expansion of epithelial cells carrying cancer-driver mutations may lead to subsequent colonization of the epithelial compartment of eutopic endometrium, and the gain of additional mutations during their development. Therefore, during menstruation, mutated endometrial cells could be delivered by retrograde flow and further proliferate. Indeed, it has been demonstrated that endometrial cells harboring somatic driver mutations frequently found in EAOC are clonally expanded in endometriosis⁴². Furthermore, the acquisition of other genetic alterations is also possible during the progression of endometriotic lesions¹⁵⁴.
Further evidence supporting this theory comes from DNA-based analysis that demonstrated clonality within region sampled from the same lesion and across different lesions¹⁴². Additionally, examinations of endometrial glands have also demonstrated notable heterogeneity in mutational profiles even among glands from the same patient⁵⁹, suggesting that different lesion may harbor distinct mutation profiles and be composed by unique cell clones.
The heterogeneity of endometriotic lesions was further demonstrated by Suda et al⁴² which demonstrated the presence of different PIK3CA mutations across multiple lesions collected from the same patient. This result supports the notion that somatic mutations in cancer driver genes may confer a selective advantage for the survival of endometrial progenitor cells and their ability to establish lesions. Additionally, vertical gland development may favor the acquisition of additional mutations. However, a similar mutational profile in multiple individual epithelial glands in the same endometriotic lesion have also been reported¹⁴². This can be explained by the presence of interconnected horizontal rhizome-like structures in the basal layer of the endometrium¹⁵⁴. Therefore, the progeny from a common progenitor epithelial cell can populate endometrial glands at distal sites. Thereby, it can be thought that the increasing heterogeneity of endometriotic glands may facilitates their potential neoplastic transformation¹⁵⁴.
Based on the reported evidence, it can be speculated that primary defect in endometriosis, and probably in EAOC can be located in the eutopic endometrium⁴²,¹⁴⁵,¹⁵⁵. Furthermore, the finding of discordant mutation profiles between paired endometriotic lesions and normal endometrial tissues in some patients⁴²,⁴⁸ could be explained by the fact that only selected clones with specific mutations may have the advantage of being able to evolve clonally and develop in ectopic sites¹¹.
This evolutionary process, in which endometrial epithelial cells with cancer-associated mutations with selective growth advantage could undergo malignant transformation in ectopic sites, have been supported by recent studies⁶⁰,¹⁵⁵.
Using whole-genome shotgun sequencing, Anglesio et al⁴⁸ analyzed seven clear cell carcinomas and targeted sequencing in synchronous endometriosis. They found multiple tumour-associated somatic mutations in at least one concurrent endometriotic lesion. Moreover, ARID1A and PIK3CA mutations appeared consistently in concurrent endometriosis when present in the primary tumor. These findings provide objective evidence that multifocal benign endometriotic lesions are clonally related and that clear cell carcinoma arising in these patients progress from endometriotic lesions that may already carry sufficient cancer-associated mutations to be considered neoplasms themselves, albeit with low malignant potential (similar to borderline tumors).
Given the high degree of conservation in mutational pattern observed in some endometriotic lesions compared to their corresponding carcinomas, and especially the occurrence of presumed driver mutations, the authors suggested that, even if somatic genetic changes in these endometriosis lesions are sufficient to allow neoplastic transformation, definitive malignant transformation is not mediated by such somatic mutations, but driven by a combination of high mutational burden, epigenetic modifiers and ovarian micro-environment⁸. Based on these results, and the presence of endometriosis lesions without the mutations present in the tumor among patients with EAOC, it can be speculated the existence of two classes of genomically distinct endometriosis respect to subsequent risk of cancer development: high-risk lesions sharing mutations with synchronous carcinoma and low-risk lesions, not sharing any mutations with the tumor⁸.
In the same line, an interesting study demonstrated the clonal lineage from normal endometrium to ovarian clear cell carcinoma through ovarian endometriosis¹⁵⁵. The authors obtained several tissue samples from a single patient with pathological diagnosis of clear cell ovarian cancer associated to endometriosis. Sampling sites included uterine endometrium, endometriotic tissue distant to the tumor, endometriotic tissue adjacent to cancer, cancer epithelial cells and cancer stromal cells. Although most of the reported mutations were shared among the different epithelial samples, demonstrating clonal relationships between spatially separated tissues, the number mutations shared with the uterine endometrium decreased from distant endometriosis to adjacent endometriosis to carcinoma. On the other hand, mutant allele frequency of the shared mutations increased sequentially from the endometrium to distant endometriosis, adjacent endometriosis and carcinoma. This pattern indicates that the genetically altered clones, including clones with cancer-driver mutations, such as PIK3CA and ARID1A, remained in a subclonal state in the endometrium and distal endometriosis, but attained a clonal state in adjacent endometriosis and the carcinoma. In the same line, previous studies have demonstrated that despite prevalent, endometrial glands harboring somatic cancer-associated mutations involved in the development of endometrial cancer remain subclonal in the endometrium with a very low progression to malignancy, probably due to non-genetic factors, such as progesterone action¹⁶¹. Therefore, in the case of endometriosis, it can be speculated that progesterone resistance could facilitate the persistence of altered clones in the endometrium. Hence, starting with a first mutation in normal endometrial glandular cells (initial genetic hit), is the sequential accumulation of genetic damage (additional genetic hits) within a continuous lineage that drives an evolving phenotype to endometriosis, and finally to ovarian cancer. In the case of EAOC, at least two genetic hits seem to be needed to achieve the biallelic ARID1A inactivation, required to trigger malignant transformation⁷¹,¹⁵⁵.
9) SOMATIC STEM CELLS AND EPITHELIAL-STROMAL INTERACTIONS IN THE DEVELOPMENT OF ENDOMETRIOSIS AND ENDOMETRIOSIS-ASSOCIATED OVARIAN CANCER
Somatic stem cells are a subset of cells residing in normal adult tissues that, through asymmetric division, retain their ability to self-renew while producing daughter cells that go on to differentiate and play a role in tissue regeneration and repair. Adult stem cells are multipotent cells capable to differentiate into several, but not all cell types (usually within the same germ lineage). The monoclonal composition of endometrial glands and the endometrial regeneration after menstruation hypothesize the existence of endometrial stem cells in the endometrium¹⁵¹,¹⁵⁴.
Recent research has implicated endometrial stem cells in the pathogenesis of endometriosis¹⁵⁴. It has been speculated that, during menstruation, circulating stem cells intended to regenerate the uterine endometrium are shed from the endometrial basal layer and may become activated and trapped outside the uterus.
An alternative hypothesis proposes that extrauterine stem/progenitor cells derived from the bone marrow-derived stem cells which home the endometrium in response to physiological injury (menstruation) and are responsible for endometrial regeneration, may differentiate into endometriotic tissue.
Current theories propose that stem cells may arise from both the epithelial and stromal layers of the endometrium, with each stem cell type playing a unique yet cooperative role in the pathogenesis of endometriosis. This model, termed the dual stem cell theory, posits that two (or more) endometrial stem/progenitor cell types collaborate to form endometriotic lesions¹⁵⁴,¹⁵⁷. To date, it is widely accepted that endometriotic epithelium and stroma are developed independently and clonally¹⁵⁸,¹⁵⁹, and several candidate cells for initiating endometriotic epithelium, and endometriotic stroma, have been identified¹⁵⁴,¹⁵⁷.
Histologically, endometriotic tissue is composed primarily of stromal cells with scant and superficially located epithelial cells. However, several studies have demonstrated that somatic mutations found in endometriosis are almost exclusively restricted to the epithelial compartment of ovarian and deep endometriotic lesions¹⁰⁵,¹⁵⁵,¹⁵⁸. Moreover, the few identified stromal mutations are not shared with epithelial samples and present at a very low mutant allele frequency¹⁶⁰. Such findings demonstrates a distinct origin of both tissues.
The presence of stem cells inside the endometrium and endometriotic lesions, and the connection between endometriosis and EAOC poses the question about the possible involvement of endometriosis stem cells in the malignant transformation of endometriosis¹⁵⁹.
Several stem cell populations have been identified in the endometrium, menstrual fluid, endometriosis and EAOC. Studies have demonstrated the expression of several stemness markers to be upregulated and highly expressed in endometriosis and endometriomas compared to eutopic endometrium and healthy controls¹⁵⁹. Furthermore, menstrual stem cells from women with endometriosis indicate increased proliferative and invasive capacity compared those from healthy women¹⁵⁹. The molecular pattern of stem cells from endometriosis lesions differs to the pattern displayed by stem cells from ectopic endometrium. On the other hand, the expression pattern of several genes and molecules responsible for the regulation of endometriotic lesions growth is similar to that expressed in EAOC¹⁵⁹.
Stroma stem cells harbor several pro-tumorigenic epigenetic marks that modulate gene expression. Epigenetic alterations in stromal cells determine local hyperestrogenism, angiogenesis, progesterone resistance and inhibitory activity on immune cells. Consequently, inflammatory factors that remodel endometrial tissues, such as PGE2 and cytokines, accumulate in the stroma¹⁶¹. This steroid-related inflammation and high proliferative activity could lead to the development of somatic driver mutations, such as ARID1A and PIK3CA, and DNA repair defects, in attached epithelial cells via paracrine signaling. Such mutations disrupt critical protein function. In years, the specific microenvironment in ovarian endometriomas may enhance the accumulation of additional mutations. Indeed, studies have shown that endometriotic implants show an increased level of DNA damage and decrease DNA repair in the course of the disease¹⁵⁹. These changes in gene expression induce phenotypic changes and proliferation in the epithelial cells, which eventually become malignant and invasive¹⁶¹. Thus, malignant transformation of endometriosis is probably related with the presence of epigenetically abnormal, poorly differentiated endometrial mesenchymal progenitor/stem cells¹⁶¹.
Considering the new paradigm of ovarian carcinogenesis³⁵,³⁶, the identification of stem cells in the tubes is an interesting finding. Paik et al¹⁶² identified epithelial stem-like cells that were concentrated in the distal part of the tube. Interestingly, these cells expressed some stemness markers also found in endometriosis stem cells. However, the importance of stem cells from tubal endometriosis in the development of EAOC remains to be elucidated.
10) EPITHELIAL-MESENCHYMAL INTERCONVERSIONS IN OVARIAN SURFACE EPITHELIUM, ENDOMETRIOSIS AND OVARIAN CANCER
Epithelial to mesenchymal transition (EMT) is a highly conserved biological process that converts immotile and polarized epithelial cells to motile mesenchymal cells. This process normally occurs during embryonic development, fibrosis, and wound healing. However, it has also been involved in cancer progression and metastasis development¹⁰³,¹⁰⁴. On the other hand, mesenchymal to epithelial transition (MET), the reverse process of EMT, is less well characterized¹⁶³.
The occurrence of such processes in different tissues is detected by the change in the corresponding protein expression marks. EMT is characterized by the gradual loss of epithelial markers (E-cadherin, cytokeratins) and the progressive gain of mesenchymal markers, such as N-cadherin, smooth-muscle actin, vimentin, fibronectin, among others. The gain of mesenchymal properties is associated with several alterations in cell functions, such as enhanced migration, invasiveness, and resistance to apoptosis¹⁰³,¹⁰⁴. However, EMT is not an all-or-nothing process, but instead a transition of epithelial to mesenchymal cells with intermediate states¹⁶⁴. Conversely, MET is characterized by the acquisition of an epithelial phenotype.
It has been hypothesized that EMT has a role in the pathogenesis of endometriosis and in the progression of ovarian cancer. Notably, in both peritoneal and ovarian endometriotic lesions the epithelial markers are downregulated, while the mesenchymal markers are upregulated¹⁰³,¹⁰⁴.
Additionally, other processes involved in endometriosis and ovarian cancer biology, such as hypoxia, ROS, and estrogens, can through different pathways to activate the EMT process. Those pathways involve many cellular factors, such as TGF-β and Wnt pathways, ultimately leading to cell proliferation and migration. For example, hypoxia induces HIF-1α, and VEGF-mediated angiogenesis, an essential process in endometriosis and ovarian cancer progression. Furthermore, ROS are found to stimulate EMT and increase invasion capacity in several human cancer cells¹⁰³,¹⁰⁴.
Estrogen-mediated signaling is involved in EMT process in endometriosis. Interestingly, estrogen-driven EMT seems to be orchestrated by the ERα. ERα directly binds to hepatocyte growth factor (HGF) and to β-catenin promoters, among other transcription factors to induce EMT in human endometrial epithelial cells¹⁰³. Additionally, HGF is a common EMT inducer in epithelial cells. HGF-activated downstream signaling molecules, such as PI3K/Akt/mTOR. HGF receptor (c-MET) have been shown to be constitutively activated and overexpressed in ovarian carcinomas¹⁰⁴.
TGF-β is involved in several aspects of endometriosis progression and tumorigenesis. Notably, TGF-β is a potent EMT inducer, and exerts its action through several signaling cascades involved in endometriosis and ovarian cancer, such as Smad, MAPK, PI3K/Akt/mTOR and Wnt/β-catenin pathways¹³⁵.
EMT process could be part of ovarian surface epithelium (OSE) physiology, involved in the postovulatory ovarian cortex repair. OSE is a stationary mesothelium with an “uncommitted” phenotype, which retains the capacity to alter its state of differentiation along stromal or epithelial phenotypes in response to environmental signals. During the postovulatory repair process, OSE cells undergo EMT and attain a fibroblast-like (mesenchymal) phenotype. In this scenario, EMT might prevent epithelial cells entrapment into the ovarian stroma during ovulation. Instead, may facilitate the entrapment of fibroblast-like stromal cells. It has been proposed that failure to undergo EMT may render the aggregation of epithelial cells within the ovarian stroma, resulting in the formation of OSE-lined inclusion cysts¹⁶⁵. Interestingly Okamoto et al¹⁶³ demonstrated the loss of mesenchymal characteristics and subsequent acquisition of epithelial characteristics, indicating MET process during inclusion cyst formation. Therefore, it is possible that inclusion cyst cells may undergo MET and subsequently differentiate into endometriosis and then undergo malignant transformation¹⁶⁶.
Although it widely accepted that these inclusion cysts are the most probable site of origin of ovarian cancer, there is no definite explanation for their formation. It has been suggested that the ovarian inclusion cysts comprise two groups of OSE cells: the first group lacking E-cadherin expression prone to EMT process during ovulatory rupture and repair process, and the second expressing E-cadherin resistant to EMT in response to ovulatory process, with a propensity to epithelial differentiation and neoplastic progression. Hence, the presence of stromal-derived factors (TGF-β, IL-6) acting in a paracrine fashion, or OSE-derived molecules (IL-1, IL-6) acting by an autocrine mechanism, may promote either EMT or neoplastic transformation of OSE within the inclusion cysts in the target cells¹⁶⁵.
In summary, EMT may have an early cytoprotective effect in ovarian function. A subtle of cells retain an epithelial phenotype and are less prone to undergo EMT in response to external stimuli. Furthermore, MET occurs during inclusion cyst formation. Such propensity towards an epithelial committed phenotype, unresponsive to normal environmental signals is consistent with the loss of normal control mechanisms characteristic of malignant transformation. Hence, it seems that persistent epithelial phenotype in the cells lining of inclusion cyst may predispose them to malignant transformation.
In this sense, it is possible to speculate that ectopic endometrial cells, which exhibit a firmly determined epithelial phenotype, could undergo malignant transformation when became trapped in an inclusion cyst.
Different carcinogenic pathways in malignant transformation of endometriosis
Malignant transformation of endometriosis is a long, complicated process involving multiple genes, signaling pathways and steps. Interplay between hormone signaling, hypoxia, inflammation and specific microenvironment within the endometriotic cyst, orchestrates transformation of endometriosis to EAOC. Many pathways converge, but at some moment they diverge exposing some differences leading to specific tumors. Despite their shared cell-of-origin and overlapping mutational profile, ovarian clear cell carcinoma and endometrioid carcinoma of the ovary may develop via different molecular, genetic and epigenetic mechanisms.
OVARIAN CLEAR CELL CARCINOMA AS A STRESS-RESPONSIVE AND EPIGENETICALLY REGULATED CANCER
Ovarian clear cell carcinoma (OCCC) has clinical characteristics and behavior that are distinct from the other epithelial ovarian tumors, including slow growth, frequent thromboembolic complications, oxidative stress tolerance, and a relative resistance to standard chemotherapy¹⁶⁷. These findings suggest that OCCC has distinct genetic background¹⁶⁸. Genetic studies have identified specifically expressed genes in OCCC which are related to the clinical characteristics of OCCC, suggesting that OCCC has a unique carcinogenic processes¹³¹. This so-called OCCC signature includes several genes and molecular networks related to oxidative stress and ROS detoxification (antioxidant defenses), hypoxia response, coagulation, glycogen metabolism, inflammation, anti-apoptosis, and chemoresistance, such as HIF-1α, IL-6, Hepatocyte nuclear factor (HNF)-1β, among others¹³¹.
Expression of OCCC signature genes is induced by treatment of immortalized OSE cells with the contents of endometriotic cysts in a time-dependent manner, indicating that the OCCC is largely dependent on the tumor microenvironment¹²⁸. Furthermore, it has been demonstrated that the expression of OCCC signature is at least in part epigenetically regulated. Yamaguchi et al¹⁶⁹ identified a distinct DNA methylation profile in OCCC compared to other subtypes of ovarian cancers. This OCCC-epigenetic profile included hypomethylation (activation) of several genes in the HNF-1 pathway, including HNF-1β gene, and synchronous hypermethylation (inactivation) of multiple genes involved in the ERα pathway, such as ES1 gene.
OCCC is characterized by unique biology and exhibits the molecular phenotypes of HNF-1 pathway activation, PI3K/Akt/mTOR activation and MAPK activation, traits that have been collectively referred to as “OCCC-likeness”¹⁶⁹. As previously mentioned, these pathways are involved in several carcinogenic networks.
HNF-1β is not only overexpressed in OCCC, but is also considered an OCCC-specific molecular marker since it is not expressed in other epithelial ovarian carcinomas (including endometrioid tumors)¹⁷⁰,¹⁷². HNF-1β has been associated with glycogen metabolism and the development of the liver, pancreas and kidney. Clinically, mutations in HNF-1β gene result in maturity-onset diabetes of the young (MODY), characterized by pancreatic hypoplasia, and kidney malformations¹⁶⁹,¹⁷³,¹⁷⁴. As OCCC is characterized by glycogen storage in the cytoplasm (causing the clear appearance) and the morphological resemblance to renal cell carcinoma, it is possible that HNF-1β may have an important role in manifesting this unique phenotype¹⁶⁹. Hence, the characteristic morphologic appearance defining OCCC seems to be, at least in part epigenetically regulated.
HNF-1β and its target genes play an important role in OCCC biology and have been implicated in the development of several unique characteristics of this tumor, such as glucose homeostasis and cell survival under hypoxia and stress conditions¹⁷¹,¹⁷³,¹⁷⁵.
Interestingly, HNF-1β is not expressed in atrophic and proliferative endometrium, OSE and ovarian inclusion cysts. On the other hand, HNF-1β expression is common in the late secretory, and menstrual endometrium.
endometrium. However, a considerable heterogeneity in HNF-1β expression is observed in epithelial cells both between and within endometriosis cases. Moreover, HNF-1β is expressed in endometriomas, but not in other benign ovarian tumors¹⁷². Kajihara et al¹⁷² demonstrated that HNF-1β expression was significantly more frequent in OCCC and adjacent atypical endometriosis than in non-atypical endometriosis. On the other hand, downregulation of HNF-1β expression was evident in endometrioid ovarian cancer and adjacent endometriosis but not in distant endometriotic lesions. Such findings rise questions about the origins of endometriosis: coelomic metaplasia for HNF-1β negative or retrograde menstruation in HNF-1 positive cases¹⁷². Indeed, it has been postulated that OCCC and endometrioid ovarian carcinoma arise from different endometriosis: HNF-1β positive and HNF-1β negative, respectively¹⁷².
Epigenetic inactivation of ES1 gene¹⁶⁹ leads to ERα downregulation. The absence of ERα has been reported in OCCC compared to other ovarian cancers¹⁷⁶. Akahane et al¹⁷⁷ showed that a gradual reduction in ERα and PR-B expression occurred with malignant transformation from endometriosis to atypical endometriosis to OCCC, suggesting that the disappearance of hormone dependency may be associated with the acquisition of the clear cell carcinoma phenotype. Interestingly, reduced ERα expression was observed in cases of endometriosis with cancer compared with cases without cancer, indicating that disappearance of hormone dependency occurs even before the development of atypical endometriosis¹⁷⁷. Although the disappearance of steroid hormone receptors seems to occur first at sites of atypical endometriosis during the progression to carcinoma, some OCCC showed direct transformation from endometriosis to OCCC in absence of atypical endometriosis¹⁷⁷. In the same line, lower detection rates of ERα, ERβ, PR-A and PR-B among OCCC compared to endometrioid ovarian cancers have been reported in other studies¹⁷⁶.
This ERα signaling-independent growth of OCCC can be attributed to epigenetic dysregulations¹⁶⁹.
Furthermore, epigenetic changes can be the earliest initiation factor and complement single driver mutations in OCCC development. The occurrence of somatic cancer-driver mutations (ARID1A and PIK3CA) could allow malignant transformation, but they cannot distinguish histologic subtypes of ovarian cancer. The specific biological characteristics of OCCC and “OCCC-likeness” are developed through coordinate alteration in DNA methylation¹⁶⁹.
Another epigenetic mechanism, histone modification, has also a role in OCCC carcinogenesis. ARID1A loss directly suppress histone deacetylase 6 (HDAC6). Yano et al¹⁷⁸ reported that HDAC6 nuclear expression was associated with immunotolerance, hypoxia resistance, and cancer stem cell phenotype acquisition in OCCC cells, leading chemoresistance and poor prognosis¹⁷⁸,¹⁷⁹. On the other hand, HDAC6 inhibition has been shown to induce apoptosis in ARID1A-inactivated OCCC cells, suggesting the potential use of HDAC6 inhibition in the treatment of OCCC¹⁷⁹.
MET is a proto-oncogene encoding MET kinase (c-MET), which is a receptor of HGF on the cell surface. HGF binds to c-MET and transmits signals downstream and activate signaling pathways promoting angiogenesis, cell proliferation and survival, such as PI3K/Akt/mTOR and MAPK networks among others¹⁷⁹. MET is expressed in epithelial cells of many organs during embryonic development and adulthood, including the liver, kidney, and bone marrow. However, MET is also activated in tumor cells leading to a series of invasive growth processes in tumorigenesis. Compared with serous ovarian cancers and normal OSE, MET amplification and expression is significantly increased in OCCC¹⁷⁷. Studies have shown that 94.2% of OCCC have high MET expression, and that MET expression intensity is closely related to chemoresistance and poor prognosis¹⁷⁹. Additionally, studies on endometriosis-associated OCCC reported that the incidence of MET overexpression increased with the malignant transformation process, from 0% in non-atypical endometriosis to 67% in atypical endometriosis to 92–100% in carcinomas¹⁷⁹. These results demonstrate that MET overexpression is an early carcinogenic event in MET-amplified OCCC and may promote the development of a subset of OCCC. The high frequency of amplification and overexpression of MET and its high specificity for OCCC have gained interest recently as an interesting possibility for targeted therapy¹⁷⁹.
In summary, OCCC phenotype acquisition is influenced by the stressful microenvironment within endometriotic cysts, including persistent exposure of epithelial cells to oxidative stress and chronic inflammation. Such carcinogenic environment leads to the transient induction of a unique gene expression (OCCC signature). Subsequently, OCCC signature becomes constitutive partly by epigenetic modifications (OCCC-specific methylation profile)¹⁸⁰.
ENDOMETRIOID OVARIAN CANCER IS A HORMONE-DEPENDENT CARCINOMA
Historically, endometrioid ovarian carcinoma (EOC) has been considered as a heterogeneous disease comprising both high-grade and low-grade tumors, with significantly different clinical behavior and prognosis. Traditionally, pathological diagnosis of EOC was largely based only on morphologic criteria.
Currently, the use of molecular markers, as well as immunohistochemical staining have showed that high proportion of the initially considered high-grade EOC were misclassified high-grade serous carcinomas (HGSC), with p53 mutations and Wilm’s tumor 1 (WT1) positivity, characteristic of type II epithelial ovarian cancer. Such misclassification was based on the presence of solid, pseudoendometrioid and/or transitional-cell-like (SET) growth pattern on histological analysis⁵¹. Consequently, high-grade endometrioid ovarian carcinomas are rare tumors not necessarily associated with the presence of endometriosis.
Endometriosis-associated endometrioid ovarian carcinomas (EOC) are characteristically low-grade. In these tumors, carcinogenesis seems to be induced by estrogen, similarly to its endometrial counterpart, and display a classical immunohistochemical profile comprising WT1 negativity (or focal positivity), wild-type tumor protein p53 (not mutated) expression, and positivity for ER and PR¹⁸¹. Akahane et al¹⁷⁷ found a gradual increase in ERα from endometriosis to atypical endometriosis to cancer in the cases of endometrioid carcinoma. In the same line, Fujimura et al¹⁷⁶ reported the expression of ERα, ERβ and PR among endometrioid endometrial cancer specimens in 100, 75 and 91.7%, respectively.
Banz et al¹ identified a group of genes involved in the regulation of autoimmunity and inflammatory cytokines expression to be specifically upregulated in endometriosis and endometriosis-associated EOC compared to benign ovaries and non-endometriosis-associated EOC. On the other hand, genes involved in cell-cell interaction, differentiation and proliferation, were equally regulated in non-endometriosis-associated EOC but not in endometriosis and benign ovaries. The authors concluded that this inflammatory molecular signature may be an important factor in the transformation of endometriosis to EOC. Interestingly, the proinflammatory role of estrogen in endometriosis has been discussed.
Endometrioid ovarian carcinomas often harbor activating mutations in the CTNNB1 gene, whereas they are exceedingly rare in OCCC⁸. Recently, Hollis et al⁸² proposed a model to stratify EOC based on their molecular profile. In this study they described distinct EOC subtypes: cases with p53 mutations, which present an aggressive clinical behavior and poor prognosis, resembling HGSC (type II tumors), and cases with CTNNB1 mutations, with low stage at diagnosis and excellent outcome. The fact that p53 and CTNNB1 mutations are mutually exclusive⁸², this model appears as a useful tool to identify low grade EOC arising from endometriosis.
ENDOMETRIOID OVARIAN AND OVARIAN CLEAR CELL CARCINOMAS ARISE FROM DIFFERENT PHASES OF THE ENDOMETRIUM
The question about how a common precursor (endometriosis) can transform into two tumors presenting striking differences in cellular phenotype and clinical behavior is intriguing. Initially it had been hypothesized that somatic mutations were responsible of such transformation. However, there is not a single recurrent mutations that is unique to either phenotype. Additionally, the frequent occurrence of such mutations in benign endometriotic lesions has clarified that they are not responsible for neoplastic progression, let alone for histotype divergence.
Previous studies have demonstrated that the deposition of epigenetic marks, such as DNA methylation, can be influenced by the transcriptional state in a given moment. Active transcription repels the DNA methylation machinery (DNMT), whereas the promoters of non-expressed genes can be easily hyper/hypomethylated, subsequently “locks” in the unexpressed state, providing mitotically heritable variation for selection during clonal evolution¹⁸⁵.
For example, in the case of OCCC, the above-described lack of transcription of ESR1 in the secretory state permits stochastic deposition of DNA methylation at this promoter. This DNA methylation gain persists through mitotic division and prevents transcriptional changes in response to estrogens¹⁸⁵.
The proliferative-endometrium-like genetic profile of ENOC is confirmed by the expression of steroid hormone receptors. On the other hand, the secretory-like phenotype of OCCC is characterized by inhibited estrogen response and high HNF-1β expression¹⁸⁵.
Conclusions
We have extensively reviewed the evolving concepts and putative mechanisms for endometriosis-associated ovarian carcinogenesis. While endometriosis is a frequent benign disease that may transform into epithelial ovarian cancer later in life, the risk of malignant transformation remains low. Evidence supporting a continuous process from endometriosis to carcinoma comes from molecular, immunohistochemical and genetic studies which have detected several common biomarkers in endometriosis (typical or atypical) and ovarian cancer.
Malignant transformation is a multistep process probably related with chronic and periodic exposure of epithelial endometriotic cells to a complex and dysregulated interplay of molecular events, involving inflammation, oxidative stress, hyperestrogenism, immunotolerance and tissue repair stimuli taking place in the unique ovarian microenvironment. Despite none of these factors seem to be sufficient alone to promote carcinogenesis, they contribute to the process in a permissive way.
To date there are not known predictive molecular markers or signs indicating which women with endometriosis are at risk of developing ovarian cancer, if they were known, active surveillance or prophylactic surgery could be indicated.
Despite several advances have been made in the understanding of endometriosis-associated ovarian carcinogenesis, the precise mechanism of neoplastic progression remains elusive.
In the future, identification of early biomarkers of malignant progression could provide the bases for targeted or immunological therapies that allow to change the historical one-size-fits-all paradigm in the management of ovarian cancer.
Conflict of Interest:
None
Acknowledgements:
None
References
1) Smolarz B, Syllo K, Romanowicz H. Endometriosis: Epidemiology, classification, pathogenesis, treatment and genetics (Review of the literature). Int J Mol Sci 2021; 22 (19): 10554. doi: 10.3390/ijms221910554.
2) Centini G, Schettini G, Pieri E, et al. Endometriosis-related ovarian cancer: Where are we now? A narrative review towards a pragmatic approach. J Clin Med 2024; 13 (7): 1933. doi: 10.3390/jcm13071933.
3) Kvaskoff M, Mahamat-Saleh Y, Farland LV, et al. Endometriosis and cancer: a systematic review and meta-analysis. Hum Reprod Update 2021; 27 (2): 393-420. doi: 10.1093/humupd/dmaa045.
4) Torre LA, Trabert B, DeSantis CE, et al. Ovarian cancer statistics, 2018. CA Cancer J Clin 2018; 68 (4): 284-96. doi: 10.3322/caac.21456.
5) Kobayashi H, Sumimoto K, Moniwa N, et al. Risk of developing ovarian cancer among women with ovarian endometrioma: a cohort study in Shizuoka, Japan. Int J Gynecol Cancer 2007; 17 (1): 37-43. doi: 10.1111/j.1525-1438.2006.00754.x.
6) Ñiguez-Sevilla I, Machado Linde F, Marín Sánchez MDP, et al. Prognostic importance of atypical endometriosis with architectural hyperplasia versus cytologic atypia in endometriosis-associated ovarian cancer. J Gynecol Oncol 2019; 30 (4): e63. doi: 10.3802/jgo.2019.30.e63.
7) McCluggage WG. Endometriosis-related pathology: a discussion of selected uncommon benign, premalignant and malignant lessions. Histopathology 2020; 76 (1): 76-92. doi: 10.1111/hi s.13970.
8) Kobayashi H, Suminoto K, Kitanaka T, et al. Ovarian endometrioma- risk factors of ovarian cancer development. Eur J Obstet Gynecol Reprod Biol 2008; 138 (2): 187-93. doi: 10.1016/j.ejogrb.2007.06.017.
9) Thomsen LH, Schnack TH, Buchardi K, et al. Risk factors of epithelial ovarian carcinomas among women with endometriosis: a systematic review. Acta Obstet Gynecol Scand 2017; 96 (6): 761-78. doi: 10.1111/aogs.13010.
10) Van Gorp T, Amant F, Neven P, Vergote I, Moerman P. Endometriosis and the development of malignant tumours of the pelvis. A review of the literature. Best Pract Res Clin Obstet Gynaecol 2004; 18 (2): 349-71. doi: 10.1016/j.bpobgyn.2003.03.001.
11) Pearce CL, Templeman C, Rossing MA, Lee A, et al. Association between endometriosis and risk of histological subtypes of ovarian cancer: a pooled analysis of case-control studies. Lancet Oncol 2012; 13 (4): 385-94. doi: 10.1016/S1470-2045(11)70404-1.
12) Hermens M, van Altena AM, Nieboer TE, et al. Incidence of endometrioid and clear-cell ovarian cancer in histological proven endometriosis: the ENOCA population-based study. Am J Obstet Gynecol 2020; 223 (1): 107.e1-107.e11. doi: 10.1016/j.ajog.2020.01.041.
13) Barnard ME, Farland LV, Yan B, Wang J, et al. Endometriosis typology and endometrial cancer risk. JAMA 2024; 332 (6): 482-9. doi: 10.1001/jam a.2024.9210.
14) Saavalainen L, Lassus H, But A, Tiitinen A, et al. Risk of gynecologic cancer according to the type of endometriosis. Obstet Gynecol 2018; 131(6): 1095-102. doi: 10.1097/AOG.0000000000 002624.
15) Mangili G, Bergamini A, Taccagni G, et al. Unraveling the two entities of endometrioid ovarian cancer: a single center clinical experience. Gynecol Oncol 2012; 126 (3): 403-7. doi: 10.1016 /j.ygyno.2012.05.007.
16) Bassiouny D, El-Baz MA, Gamil TM, Shams N, et al. Endometriosis-associated ovarian cancer is a subset with a more favorable outcome and distinct clinical-pathologic characteristics. Int J Gynecol Pathol 2019; 38 (5): 435-42. doi: 10.1097 /PGP.0000000000000533.
17) Wang S, Qiu L, Lang JH, et al. Clinical analysis of ovarian epithelial carcinoma with coexisting pelvic endometriosis. Am J Obstet Gynecol 2013; 208 (5): 413.e1-5. doi: 10.1016/j.ajog.2012.12.004.
18) Kumar S, Munkarah A, Arabi H, et al. Prognosis analysis of ovarian cancer associated with endometriosis. Am J Obstet Gynecol 2011; 204 (1): 63.e1-7. doi: 10.1016/j.ajog.2010.08.017.
19) Bounous V, Ferrero A, Fuso L, Ravarino N, et al. Endometriosis-associated ovarian cancer: a distinct clinical entity? Anticancer Res 2016; 36 (7): 3445-9.
20) Davis M, Rauh-Hain JA, Andrade C, et al. Comparison of clinical outcomes of patients with clear cell and endometrioid ovarian cancer associated with endometriosis to papillary serous carcinoma of the ovary. Gynecol Oncol 2014; 132 (3): 760-6. doi: 10.1016/j.ygyno.2014.01.012.
21) Ju UC, Kang WD, Kim SM. The effect of concurrent endometriosis on the prognosis of women with ovarian clear cell or endometrioid carcinoma. Int Gynaecol Obstet 2019; 146 (2): 177-83. doi: 10.1002/ijgo.12861.
22) Noli S, Cipriani S, Scarfone G, et al. Long term survival of endometriosis associated clear cell and endometrioid ovarian cancers. Int J Gynecol Cancer 2013; 23 (2): 244-8. doi: 10.1097/IGC.0 b013e31827aa0bb.
23) Cuff J, Longacre TA. Endometriosis does not confer prognosis in ovarian carcinoma of uniform cell type. Am J Surg Pathol 2012; 36 (5):688-95. doi: 10.1097/PAS.0b013e31824b6eed.
24) Sampson JA. Endometrial carcinoma of the ovary, arising in endometrial tissue in that organ. Arch Surg 1925; 10 (1): 1-72. doi:10.1001/archsurg.1925.01120100007001.
25) Scott RB. Malignant changes in endometriosis. Obstet Gynecol 1953; 2 (3): 283-9.
26) Capozzi VA, Scarpelli E, dell`Omo S, Rolla M, et al. Atypical endometriosis: A comprehensive systematic review of pathological patterns and diagnostic challenges. Biomedicines 2024; 12 (6): 1209. doi: 10.3390/biomedicines12061209.
27) Fukunaga M, Nomura K, Ishikawa E, et al. Ovarian atypical endometriosis: its close association with malignant epithelial tumors. Histopathology 1997; 30 (3): 249-55. doi: 10.1046/j.1365-2559.199 7.d01-592.x.
28) Guidozzi F. Endometriosis-associated cancer. Climateric 2021; 24 (6): 587-92. doi: 10.1080/1369 7137.2021.1948994.
29) Del Mundo MM, Aguilar M, Chen H, et al. β-catenin, PAX2, and PTEN aberrancy across the spectrum of endometrioid ovarian lesions. Int J Gynecol Pathol 2024. doi: 10.1097/PGP.00000000 00001046. Online ahead of print.
30) Wepy C, Nucci MR, Parra-Herran. Atypical endometriosis: Comprehensive characterization of clinic-pathologic, immunohistochemical, and molecular features. Int J Gynecol Pathol 2023; 43 (1): 70-7. doi: 10.1097/PGP.0000000000000952.
31) Fukunaga M, Ushigome S. Epithelial metaplastic changes in ovarian endometriosis. Mod Pathol 1998; 11(8): 784-8.
32) Ogawa S, Kaku T, Amada S, et al. Ovarian endometriosis associated with ovarian carcinoma: a clinicopathological and immunohistochemical study. Gynecol Oncol 2000; 77 (2) : 298-304. doi: 10.1006/gyno.2000.5765.
33) Yamamoto S, Tsuda H, Miyai K, Takano M, Tamai S, Matsubara O. Cumulative alterations in p27-related cell-cycle regulators in the development of endometriosis-associated ovarian clear cell adenocarcinoma. Histopathology 2010; 56 (6): 740-9. doi: 10.1111/j.1365-2559.2010.03551.x.
34) Vercellini P, Cribiù FM, Del Gobbo A, Carcangiu ML, Somigliana E, Bosari S. The oncofetal protein IM3: a novel biomarker and triage tool for premalignant atypical endometriotic lesions. Fertil Steril 2013; 99 (7): 1974-9. doi: 10.1016/j.fertnstert.2013.02.002.
35) Kurman RJ, Shih IE. The origin and pathogenesis of epithelial ovarian cancer: a proposed unifying theory. Am J Surg Pathol 2010; 34 (3): 433-43. doi: 10.1097/PAS.0b013e3181cf3d79.
36) Kurman RJ, Shih IE. The dualistic model of ovarian carcinogenesis: revisited, revised and expanded. Am J Pathol 2016; 186 (4): 733-47. doi: 10.1016/j.ajpath.2015.11.011.
37) Hollis RL. Molecular characteristics and clinical behaviour of epithelial ovarian cancers. Cancer Lett 2023; 555: 216057. doi: 10.1016/j.ca nlet.2023.216057.
38) Taylor HS, Kotlyar AM, Flores VA. Endometriosis is a chronic systemic disease: clinical challenges and novel innovations. Lancet 2021; 397 (10276): 839-52. doi: 10.1016/S0140-6736(21) 00389-5.
39) Gazvani R, Templeton A. New consideration for the pathogenesis of endometriosis. Int J Gynaecol Obstet 2002; 76 (2): 117-26. doi: 10.101 6/s0020-7292(01)00577-x.
40) Sampson JA. Peritoneal endometriosis due to the menstrual dissemination of endometrial tissue into the peritoneal cavity. Am J Obstet Gynecol 1927; 14 (4): 442-69. https://doi.org/10.1016/s0002-9378(15)30003-x
41) Vercellini P, Crosignani P, Somigliana E, et al. The “incessant menstruation” hypothesis: a mechanistic ovarian cancer model with implications for prevention. Hum Reprod 2011; 26 (9): 2262-73. doi: 10.1093/humrep/der211.
42) Suda K, Nakaoka H, Yoshihara K, et al. Clonal expansion and diversification of cancer-associated mutations in endometriosis and normal endometrium. Cell Reports 2018; 24 (7): 1777-89. doi: 10.1016/j.celrep.2018.07.037.
43) Guo SW. Cancer-associated mutations in endometriosis: shedding light on the pathogenesis and pathophysiology. Hum Reprod Update 2020; 26 (3): 423-49. doi: 10.1093/humupd/dmz047.
44) Cadish LA, Sheperd JP, Barber EL, Ridgeway B. Risks and benefits of opportunistic salpingectomy during vaginal hysterectomy: a decision-analysis. Am J Obstet Gynecol 2017; 217 (5): 603.e1-6. doi: 10.1016/j.ajog.2017.06.007.
45) Modugno F, Ness RB, Allen GO, Schildkraut JM, Davis FG, Goodman MT. Oral contraceptive use, reproductive history, and risk of epithelial ovarian cancer in women with and without endometriosis. Am J Obstet Gynecol 2004; 191 (3): 733-40. doi: 10.1016/j.ajog.2004.03.035.
46) Dinulescu DM, Ince TA, Quade BJ, Shafer SA, Crowley D, Jacks T. Role of K-ras and Pten in the development of mouse models of endometriosis and endometrioid ovarian cancer. Nat Med 2005; 11 (1): 63-70. doi: 10.1038/nm1173.
47) Cibula D, Widschwendter M, Májek O, Dusek L. Tubal ligation and the risk of ovarian cancer: review and meta-analysis. Hum Reprod Update 2011; 17 (1): 55-67. doi: 10.1093/humupd/ dmq030.
48) Tworoger SS, Fairfield KM, Colditz GA, Rosner BA, Hankinson SE. Association of oral contraceptive use, other contraceptive methods, and infertility with ovarian cancer risk. Am J Epidemiol 2007; 166 (8): 894-901. doi: 10.1093/aje/kwm157.
49) Riman T, Nilsson S, Perrson IR. Review of epidemiological evidence for reproductive and hormonal factors in relation to the risk of epithelial ovarian malignancies. Acta Obstet Gynecol Scand 2004; 83 (9): 783-95. doi: 10.1111/j.0001-6349.2 004.00550.x.
50) Fathalla M. Incessant ovulation – a factor in ovarian neoplasia? Lancet 1971; 2 (7716): 163. doi: 10.1016/s0140-6736(71)92335-x.
51) Katabushi H, Okamura H. Cell biology of human ovarian surface epithelial cells and ovarian carcinogenesis. Med Electron Microsc 2003; 36 (2): 74-86. doi: 10.1007/s00795-002-0196-6.
52) Collaborative Group on Epidemiological Studies of Ovarian Cancer, Beral V, Doll R, Hermon C, Peto R, Reeves G. Ovarian cancer and oral contraceptives: collaborative reanalysis of data from 45 epidemiological studies including 23,257 women with ovarian cancer and 87,303 controls. Lancet 2008; 371 (9609): 303-14. doi: 10.1016/S0 140-6736(08)60167-1.
53) Prodromidou A, Kathopoulis N, Zacharakis D, Grigodiaris T, Chatzipapas I, Protopapas A. Tubal endometriosis: from bench to bedside, a review. J Pers Med 2022: 12 (3): 362. doi: 10.33 90/jpm12030362.
54) Hermens M, van Altena AM, Bulten J, Siebers AG, Bekkers RLM. Increased association of ovarian cancer in women with histological proven endosalpingiosis. Cancer Epidemiol 2020; 65: 101700. doi: 10.1016/j.canep.2020.101700.
55) Lac V, Nazeran TM, Tessier-Cloutier B, et al. Oncogenic mutations in histologically normal endometrium: the new normal? J Pathol 2019; 249 (2): 173-81. doi: 10.1002/path.5314.
56) Madanes D, Bilotas MA, Bastón JI, et al. PI3K/AKT pathway is altered in the endometriosis patient`s endometrium and presents differences according to severity stage. Gynecol Endocrinol 2020; 36 (5): 436-40. doi: 10.1080/09513590.201 9.1680627.
57) Wu Y, Kajdacsy-Balla A, Strawn E, et al. Transcriptional characterizations of differences between eutopic and ectopic endometrium. Endocrinology 2006; 147 (1): 232-46. doi: 10.1210/ en.2005-0426.
58) Li X, Zhang Y, Zhao L, et al. Whole-exome sequencing of endometriosis identifies frequent alterations in genes involved in cell adhesion and chromatin-remodeling complexes. Hum Mol Genet 2014; 23 (22): 6008-21. doi: 10.1093/hmg/ddu330.
59) Moore L, Cagan A, Coorens THH, et al. The mutational landscape of normal human endometrial epithelium. Nature 2020; 580 (7805): 640-6. doi: 10.1038/s41586-020-2214-z.
60) Anglesio MS, Papadopoulos N, Ayhan A, et al. Cancer-associated mutations in endometriosis without cancer. N Engl J Med 2017; 376 (19): 1835-48. doi: 10.1056/NEJMoa1614814.
61) Wiegan KC, Shah SP, Al-Agha OM, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med 2010; 363 (16): 1532-43. doi: 10.1056/NEJMoa1008433.
62) Jones S, Wang T-L, Shih I-M, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 2010; 330 (6001): 228-31. doi: 10.1126/science.1196333.
63) Govatati S, Kodati VL, Deenadayal M, Chakravarty B, Shivaji S, Bhanoori M. Mutations in PTEN tumor gene and risk of endometriosis: a case-control study. Hum Reprod 2014; 29 (2): 324-36. doi: 10.1093/humrep/det387.
64) Sato N, Tsunoda H, Nishida M, et al. Loss of heterozygosity on 10q23.3 and mutation of the tumor suppressor gene PTEN in benign endometrial cyst of the ovary: possible sequence progression from benign endometriosis cyst to endometrioid carcinoma and clear cell carcinoma of the ovary. Cancer Res 2000; 60 (24): 7052-6.
65) Koppolu A, Maksym RB, Paskal W, et al. Epithelial cells of deep infiltrating endometriosis harbor mutations in cancer driver genes. Cells 2021; 10 (4): 749. doi: 10.3390/cells10040749.
66) Samartzis EP, Samartis N, Noske A, et al. Loss of ARID1A/BAF250a-expression in endometriosis: a biomarker for risk of carcinogenic transformation? Mod Pathol 2012; 25 (6): 885-92. doi: 10.1038/modpathol.2011.217.
67) Zou Y, Zhou J-Y, Guo J-B, et al. The presence of KRAS, PPP2R1A and ARID1A mutations in 101 Chinese samples with ovarian endometriosis. Mutat Res 2018; 809: 1-5. doi: 10.1016/j.mrfmmm.2018.0 3.001.
68) Anglesio MS, Bashashati A, Wang YK, et al Multifocal endometriotic lesions associated with cancer are clonal and carry high mutation burden. J Pathol 2015; 236 (2): 201-9. doi: 10.1002/path.4516.
69) Stamp JP, Gilks CB, Wesseling M, et al. BAF250a expression in atypical endometriosis and endometriosis-associated ovarian cancer. Int J Gynecol Cancer 2016; 26 (5): 825-32. doi: 10.1097 /IGC.0000000000000698.
70) Er TK, Su YF, Wu CC, et al. Targeted next-generation sequencing for molecular diagnosis of endometriosis-associated ovarian cancer. J Mol Med (Berl) 2016; 94 (7): 835-47. doi: 10.1007/s0 0109-016-1395-2.
71) Banz C, Ungethuem U, Kuban RJ, Diedrich K, Lengyel E, Hornung D. The molecular signature of endometriosis-associated endometrioid ovarian cancer differs significantly from endometriosis-independent endometrioid ovarian cancer. Fertil Steril 2010; 94 (4): 1212-7. doi: 10.1016/j.fertnster t.2009.06.039.
72) Samartzis E, Noske A, Dedes KJ, Fink D, Imesch P . ARID1A mutations and PI3K/AKT pathway alterations in endometriosis and endometriosis-associated ovarian carcinomas. Int J Mol Sci 2013; 14 (9): 18824-49. doi: 10.3390/ijms1 40918824.
73) Wu RC, Wang TL, Shih IeM. The emerging roles of ARID1A in tumor suppression. Cancer Biol Ther 2014: 15 (6): 655-64. doi: 10.4161/cbt.28411.
74) Xiao W, Awadallah A, Xin W. Loss of ARID1A/BAF250a expression in ovarian endometriosis and clear cell carcinoma. Int J Clin Exp Pathol 2012; 5 (7): 642-50.
75) Chene G, Ouellet V, Rahimi K, Barres V, Provencher D, Mes-Masson AM. The ARID1A pathway in ovarian clear cell and endometrioid carcinoma, contiguous endometriosis, and benign endometriosis. Int J Gynaecol Obstet 2015; 130 (1): 27-30.
76) Ayhan A, Mao TL, Seckin T, et al. Loss of ARID1A expression is an early molecular event in tumor progression from ovarian endometriotic cyst to clear cell and endometrioid carcinoma. Int J Gynecol Cancer 2012; 22 (8): 1310-5. doi: 10.109 7/IGC.0b013e31826b5dcc.
77) Yachida N, Yoshihara K, Suda K, et al. ARID1A protein expression is retained in ovarian endometriosis with ARID1A loss-of-function mutations: Implication for the “two-hit” hypothesis. Sci Rep 2020; 10 (1): 14260. doi: 10.1038/s41598-020-71273-7.
78) Lakshminarasimhan R, Andreu-Vieyra C, Lawrenson K, et al. Down-regulation of ARID1A is sufficient to initiate neoplastic transformation along with epigenetic reprogramming in non-tumorigenic endometriotic cells. Cancer Lett 2017; 401: 11-19. doi: 10.1016/j.canlet.2017.04.040.
79) Wu CH, Mao TL, Vang R, et al. Endocervical-type mucinous borderline tumors are related to endometrioid based on mutation and loss of expression of ARID1A. Int J Gynecol Pathol 2012; 31 (4): 297-303. doi: 10.1097/PGP.0b013e31823f 8482.
80) McCluggage G, Singh N, Gilks B. Key changes to the World Health Organization (WHO) classification of female genital tumours introduced in the 5th edition (2020). Histopathology 2022; 80 (5): 762-78. doi: 10.1111/his.14609.
81) Hu Y, Fu K, Liu H, et al. Ovarian seromucinous carcinoma: an independent epithelial ovarian cancer? J Ovarian Res 2023; 16 (1): 18. doi: 10.118 6/s13048-023-01100-w.
82) Mikhaleva LM, Davydov AI, Patsap OI, et al. Malignant transformation and associated biomarkers of ovarian endometriosis: a narrative review. Adv Ther 2020; 37 (6): 2580-603. doi: 10.1007/s12325-020-01363-5.
83) Tang L, Bian C. Recent progress in endometriosis-associated ovarian cancer. Front Oncol 2024; 14: 1381244. doi: 10.3389/fonc.2024.1381244.
84) Guan B, Rahmanto YS, Wu RC, Wang Y, Wang Z, Wang TL, Shih IeM. Roles of deletion of Arid1a, a tumor suppressor, in mouse ovarian tumorigenesis. J Natl Cancer Inst 2014; 106 (7): dju146. doi: 10.1093/jnci/dju146.
85) Huang HN, Lin MC, Huang WC, Chiang YC, Kuo KT. Loss of ARID1A expression and its relationships with PI3K-Akt pathway alterations and ZNF217 amplification in ovarian clear cell carcinoma. Mod Pathol 2014; 27 (7): 983-90. doi: 10.1038/mo dpathol.2013.216.
86) Driva TS, Schatz C, Haybaeck J. Endometriosis-associated ovarian carcinomas: How a PI3K/AKT/mTOR pathway affects their pathogenesis. Biomolecules 2023; 13 (8): 1253. doi: 10.3390/biom13081253.
87) Chandler RL, Damrauer JS, Raab JR, et al. Coexistent ARD1A-PIK3CA mutations promotes ovarian clear-cell tumorigenesis through pro-tumorigenic inflammatory cytokine signaling. Nat Commun 2015; 6: 6118. doi: 10.1038/ncomms7118.
88) Luchini C, Veronese N, Solmi M, et al. Prognostic role and implications of mutation status of tumor suppressor gene ARID1A in cancer: a systematic review and meta-analysis. Oncotarget 2015; 6 (36): 39088-97. doi: 10.18632/oncotarg et.5142.
89) Heinze K, Nazeran TM, Lee S, et al. Validated biomarker assays confirm that ARID1A loss is confounded with MMR deficiency, CD8+ TIL infiltration, and provides no independent prognostic value in endometriosis-associated ovarian carcinomas. J Pathol 2022; 256 (4): 388-401. doi: 10.1002/path.5849.
90) Kato M, Masashi T, Miyamoto M, et al. Effect of ARID1A/BAF250a expresión on carcinogenesis and clinicopathological factors in pure-type clear cell adenocarcinoma of the ovary. Mol Clin Oncol 2016; 5 (4): 395-401. doi: 10.38 92/mco.2016.973.
91) Campbell IG, Russel SE, Choong DY, et al. Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res 2004; 64 (21): 7678-81. doi: 10.1158/0008-5472.CAN-04-2933.
92) Yamamoto S, Tsuda H, Takano M, Iwaya K, Tamai S, Matsubara O. PIK3CA mutations is an early event in the development of endometriosis-associated ovarian clear cell adenocarcinoma. J Pathol 2011; 225 (2): 189-94. doi: 10.1002/path.2 940.
93) Kuo KT, Mao TL, Jones S, et al. Frequent activating mutations of PIK3CA in ovarian clear cell carcinoma. Am J Pathol 2009; 174 (5): 1597-601. doi: 10.2353/ajpath.2009.081000.
94) Sekizawa A, Amemiya J, Otsuka J, et al, Malignant transformation of endometriosis: application of laser microdissection for analysis of genetic alterations according to pathological changes. Med Electron Microsc 2004; 37 (2): 97-100. doi: 10.1007/s00795-003-0233-0.
95) Wu R, Hendrix-Lucas N, Kuick R, et al. Mouse model of human ovarian endometrioid adenocarcinoma based on somatic defects in the Wnt/-catenin and PI3K/Pten signaling pathways. Cancer Cell 2007; 11 (4): 321-33. doi: 10.1016/j.c cr.2007.02.016.
96) Aziz AUR, Farid S, Qin K, Wang H, Liu B. PIM kinases and their relevance to the PI3K/AKT/ mTOR pathway in the regulation of ovarian cancer. Biomolecules 2018; 8 (1): 7. doi: 10.3390/biom8 010007.
97) Ediriweera MK, Tennekoon KH, Samarakoon SR. Role of the PI3K/AKT/mTOR signaling pathway in ovarian cancer: Biological and therapeutic significance. Semin Cancer Biol 2019; 59: 147-60. doi: 10.1016/j.semcancer.2019.05.012.
98) Arend RC, Londoño-Joshi AI, Straughn JM Jr, Buchsbaum DJ. The Wnt/-catenin pathway in ovarian cancer: A review. Gynecol Oncol 2013; 131 (3): 772-9. doi: 10.1016/j.ygyno.2013.09.034.
99) Kiewisz J, Wasniewski T, Kmiec Z. Participation of WNT and -catenin in physiological and pathological endometrial changes: association with angiogenesis. Biomed Res Int 2015; 2015: 854056. doi: 10.1155/2015/854056.
100) Klemmt PAB, Starzinski-Powitz A. Molecular and cellular pathogenesis of endometriosis. Curr Womens Health Rev 2018; 14 (2): 106-16. doi: 10.2174/1573404813666170306163448.
101) Zhang M, Xu T, Tong D, et al. Research advances in endometriosis-related signaling pathways: A review. Biomed Pharmacother 2023; 164: 114909. doi: 10.1016/j.biopha.2023.114909.
102) Zhang Y, Sun X, Li Z, et al. Interactions between miRNAs and the Wnt/-catenin signaling pathway in endometriosis. Biomed Pharmacother 2024; 171: 116182. doi: 10.1016/j.biopha.2024.1 16182.
103) Yang YM, Yang WX. Epithelial-to-mesenchymal transition in the development of endometriosis. Oncotarget 2017; 8 (25): 41679-89. doi: 10.18632/oncotarget.16472.
104) Vergara D, Merlot B, Lucot JP, et al. Epithelial-mesenchymal transition in ovarian cancer. Cancer Lett 2010; 291(1): 59-66. doi: 10.1016/j.canlet.2009.09.017.
105) Linder A, Westbom-Fremer S, Mateoiu C, et al. Genomic alterations in ovarian endometriosis and subsequently diagnosed ovarian carcinoma. Hum Reprod 2024; 39 (5): 1141-54. doi: 10.109 3/humrep/deae043.
106) Calmon MS, Lemos FFB, Silva Luz , et al. Immune pathway through endometriosis to ovarian cancer. World J Clin Oncol 2024: 15 (4): 496-522. doi: 10.5306/wjco.v15.i4.496.
107) Yunna C, Mengru H, Lei W, Weidong C. Macrophage M1/M2 polarization. Eur J Pharmacol 2020; 877: 173090. doi: 10.1016/j.ejphar.2020.17 3090.
108) Strizova Z, Benesova I, Bartolini R, et al. M1/M2 macrophages and their overlaps – myth or reality? Clin Sci (Lond) 2023; 137 (15): 1067-93. doi: 10.1042/CS20220531.
109) Brunty S, Clower L, Mitchell B, Fleshman T, Zgheib NB, Santanam N. Peritoneal modulators of endometriosis-associated ovarian cancer. Front Oncol 2021; 11: 793297. doi: 10.3389/fonc.202 1.793297.
110) Szlosarek PW, Grimshaw MJ, Kulbe H, et al. Expression and regulation of tumor necrosis factor in normal and malignant ovarian epithelium. Mol Cancer Ther 2006; 5 (2): 382-90. doi: 10.1158/153 5-7163.MCT-05-0303.
111) Yang HL, Zhou WJ, Chan KK, et al. The crosstalk between endometrial stromal cells and macrophages impairs cytotoxicity of NK cells in endometriosis by secreting IL-10 and TGF-. Reproduction 2017; 154 (6): 815-25. doi: 10.1530 /REP-17-0342.
112) Kang YJ, Jeung IC, Park A, et al. An increased level of IL-6 suppresses NK cell activity in peritoneal fluid of patients with endometriosis via regulation of SHP-2 expression. Hum Reprod 2014; 29 (10): 2176-89. doi: 10.1093/humrep/deu172.
113) Sikora J, Smycz-Kubanska M, Mielczarek-Palacz A, Bednarek I, Kondera-Anasz Z. The involvement of multifuncional TGF-beta and related cytokines in pathogenesis of endometriosis. Immunol Lett 2018; 201: 31-7. doi: 10.1016/j.imlet. 2018.10.011.
114) Ibrahim MG, Sillem M, Plendl J, et al. Arrangement of myofibroblastic and smooth muscle-like cells in superficial peritoneal endometriosis and a possible role of transforming growth factor beta 1 (TGF1) in myofibroblastic metaplasia. Arch Gynecol Obstet 2019; 299 (2): 489-99. doi: 10.10 07/s00404-018-4995-y.
115) Nilsson EE, Skinner MK. Role of transforming growth factor in ovarian surface epithelium biology and ovarian cancer. Reprod Biomed Online 2002; 5 (3): 254-8. doi: 10.1016/s1472-6483(10)6 1828-7.
116) Mikula-Pietrasik J, Rutecki S, Ksiazek K. The functional multipotency of transforming growth factor signaling at the intersection of senescence and cancer. Cell Mol Life Sci 2022; 79 (4): 196. doi: 10.1007/s00018-022-04236-y.
117) Young VJ, Brown JK, Saunders PT, Duncan WC, Horne AW. The peritoneum is both a source and target of TGF- in women with endometriosis. PLoS One 2014; 9 (9): e106773. doi: 10.1371/journ al.pone.0106773.
118) Young VJ, Ahmad SF, Duncan WC, Horne AW. The role of TGF- in the pathophysiology of peritoneal endometriosis. Hum Reprod Update 2017; 23 (5): 548-59. doi: 10.1093/humupd/dmx016.
119) Liu YG, Tekmal RR, Binkley PA, Nair HB, Schenken RS, Kirma NB. Induction of epitelial cell invasion and c-fms expression by transforming growth factor beta. Mol Hum Reprod 2009; 15 (10): 665-73. doi: 10.1093/molehr/gap043.
120) Carli C, Metz CN, Al-Abed Y, Naccache PH, Akoum A. Up-regulation of cyclooxigenase-2 expression and prostaglandin E2 production in human endometriotic cells by macrophage migration inhibitory factor: involvement of a novel kinase signaling pathway. Endocrinology 2009; 150 (7): 3128-37. doi: 10.1210/en.2008-1088.
121) Choi HJ, Park MJ, Kim BS, et al. Transforming growth factor 1 enhances adhesion of endometrial cells to mesothelium by regulating integrin expression. BMB Rep 2017; 50 (8): 429-34. doi: 10.5483/bmbrep.2017.50.8.097.
122) Starzinski-Powitz A, Handrow-Metzmacher H, Kotzian S. The putative role of cell adhesion molecules in endometriosis: can we learn from tumor metastasis? Mol Med Today 1999; 5 (7): 304-9. doi: 10.1016/s1357-4310(99)01497-5.
123) Jeong SK, Kim JS, Lee CG, et al. Tumor associated macrophages provide the survival resistance of tumor cells to hypoxic microenvironmental condition through IL-6 receptor-mediated signals. Immunobiology 2017; 222 (1): 55-65. doi: 10.1016/j.imbio.2015.11.010.
124) Mao CL, Seow KM, Chen KH. The utilization of bevacizumab in patients with advanced ovarian cancer: a systematic review of the mechanisms and effects. Int J Mol Sci 2022; 23 (13): 6911. doi: 10.3390/ijms23136911.
125) Guo Y, Xu F, Lu T, Duan Z, Zhang Z. Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat Rev 2012; 38 (7): 904-10. doi: 10.1016/j.ctrv.2012.04.007.
126) Suryawanshi S, Huang X, Elishaev E, et al. Complement pathway is frequently altered in endometriosis and endometriosis-associated ovarian cancer. Clin Cancer Res 2014; 20 (23): 6163-74. doi: 10.1158/1078-0432.CCR-14-1338.
127) Kobayashi H. Potential scenarios leading to ovarian cancer arising from endometriosis. Redox Rep 2016; 21(3): 119-26. doi: 10.1179/135100021 5Y.0000000038.
128) Yamaguchi K, Mandai M, Toyokuni S, et al. Contents of endometriotic cysts, especially the high concentrations of free iron, are a possible cause of carcinogenesis in the cysts through the iron-induced persistent oxidative stress. Clin Cancer Res 2008; 14 (1): 32-40. doi: 10.1158/1078-0432.CCR-07-1614.
129) Scutiero G, Iannone P, Bernardi G, et al. Oxidative stress and endometriosis: A systematic review of the literature. Oxid Med Cell Longev 2017; 2017: 7265238. doi: 10.1155/2017/7265238.
130) Steinbuch SC, LüB AM, Eltrop S, Götte M, Kiesel L. Endometriosis-associated ovarian cancer: From molecular pathologies to clinical relevance. Int J Mol Sci 2024; 25 (8): 4306. doi: 10.3390/ijm s25084306.
131) Yamaguchi K, Mandai M, Oura T, et al. Identification of an ovarian clear cell carcinoma gene signature that reflects inherent disease biology and the carcinogenic processes. Oncogene 2010; 29 (12): 1741-52. doi: 10.1038/onc.2009.470.
132) Kajiyama H, Suzuki S, Yoshihara M, et al. Endometriosis and cancer. Free Radic Biol Med 2019; 133: 186-92. doi: 10.1016/j.freeradbiomed.2 018.12.015.
133) Zanetta G, Webb MJ, Li H, Keeney GL. Hyperestrogenism: a relevant risk factor for the development of cancer from endometriosis. Gynecol Oncol 2000; 79 (1): 18-22. doi: 10.1006/gyno.200 0.5905.
134) Marino M, Galluzzo P, Ascenzi P. Estrogen signaling pathways to impact gene transcription. Curr Genomics 2006: 7 (8): 497-508. doi: 10.217 4/138920206779315737.
135) Zhang L, Xiong W, Xiong Y, Liu H, Liu Y. 17 -Estradiol promotes vascular endothelial growth factor expression via the Wnt/-catenin pathway during the pathogenesis of endometriosis. Mol Hum Reprod 2016; 22 (7): 526-35. doi: 10.1093/mo lehr/gaw025.
136) Andersen CL, Boisen MM, Sikora MJ, et al. The evolution of estrogen receptor signaling in the progression of endometriosis to endometriosis-associated ovarian cancer. Horm Cancer 2018; 9 (6): 399-407. doi: 10.1007/s12672-018-0350-9.
137) Nasu K, Kawano Y, Tsukamoto Y, et al. Aberrant DNA methylation status of endometriosis: epigenetics as the pathogenesis, biomarker and therapeutic agent. J Obstet Gynaecol Res 2011; 37 (7): 683-95. doi: 10.1111/j.1447-0756.2011.01663.x.
138) Munsksgaard PS, Blaakaer J. The association between endometriosis and ovarian cancer: a review of histological, genetic and molecular alterations. Gynecol Oncol 2012; 124 (1): 164-9. doi: 10.1016/j.ygyno.2011.10.001.
139) Rossi M, Seidita I, Vannuccini S, Prisinzano M, Donati C, Petraglia F. Epigenetics, endometriosis and sex steroid receptors: an update on the epigenetic regulatory mechanisms of estrogen and progesterone receptors in patients with endometriosis. Vitam Horm 2023; 122: 171-91. doi: 10.1016/bs.vh.2023.01.007.
140) Monsivais D, Dyson MT, Yin P, et al. ER-- and prostaglandin E2-regulated pathways integrate cell proliferation via Ras-like and estrogen-regulated growth inhibitor in endometriosis. Mol Endocrinol 2014; 28 (8): 1304-15. doi: 10.1210/me.2013-1421.
141) Guo C, Ren F, Wang D, et al. RUNX3 is inactivated by promoter hypermethylation in malignant transformation of ovarian endometriosis. Oncol Rep 2014; 32 (6): 2580-8. doi: 10.3892/or.2 014.3524.
142) Guo SW. Epigenetics of endometriosis. Mol Hum Reprod 2009; 15 (10): 587-607. doi: 10.109 3/molehr/gap064.
143) Yotova I, Hsu E, Do C, et al. Epigenetic alterations affecting transcription factors and signaling pathways in stromal cells of endometriosis. PLoS One 2017; 12 (1): e0170859. doi: 10.1371/journa l.pone.0170859.
144) He J, Chang W, Feng C, Cui M, Xu T. Endometriosis malignant transformation: Epigenetics as a probable mechanism in ovarian tumorigenesis. Int J Genomics 2018; 2018: 1465348. doi: 10.115 5/2018/1465348.
145) Brunty S, Mitchell B, Bou-Zgheib, Santanam N. Endometriosis and ovarian cancer risk, an epigenetic connection. Ann Transl Med 2020; 8 (24): 1715. doi: 10.21037/atm-20-2449.
146) Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol 2010; 28 (10): 1057-68. doi: 10.1038/nbt.1685.
147) Wang D, Guo C, Li Y, et al. Oestrogen up-regulates DNMT1 and leads to hypermethylation of RUNX3 in the malignant transformation of ovarian endometriosis. Reprod Biomed Online 2022; 44 (1): 27-37. doi: 10.1016/j.rbmo.2021.06.030.
148) Wu Y, Strawn E, Starzinski-Powitz A, Guo SW. Prolonged stimulation with tumor necrosis factor- induced partial methylation at PR-B promoter in immortalized epithelial-like endometriotic cells. Fertil Steril 2008; 90 (1): 234-7. doi: 10.1016/j.fe rtnstert.2007.06.008.
149) Xie H, Chen P, Huang HW, Liu LP, Zhao F. Reactive oxygen species downregulate ARID1A expression via its promoter methylation during the pathogenesis of endometriosis. Eur Rev Med Pharmacol Sci 2014; 21 (20): 4509-15.
150) Gaia-Oltean AI, Braicu C, Gulei D, et al. Ovarian endometriosis, a precursor of ovarian cancer: Histological aspects, gene expression and microRNA alterations (Review). Exp Ther Med 2021; 21 (3): 243. doi: 10.3892/etm.2021.9674.
151) Kyo S, Sato S, Nakayama K. Cancer-associated mutations in normal endometrium: Surprice or expected? Cancer Sci 2020; 111 (10): 3458-67. doi: 10.1111/cas.14571.
152) Wu Y, Basir Z, Kajdacsy-Balla A, et al. Resolution of clonal origins for endometriotic lesions using laser capture microdissection and the human androgen receptor (HUMARA) assay. Fertil Steril 2003; 79 suppl 1: 710-7. doi: 10.1016/s0015-0282(02)04821-5.
153) Yano T, Jimbo H, Yoshikawa H, Tsutsumi O, Taketani Y. Molecular analysis of clonality in ovarian endometrial cysts. Gynecol Obstet Invest 1999; 47 suppl 1: 41-5. doi: 10.1159/000052858.
154) Adilvayeva A, Kunz J. Pathogenesis of endometriosis and endometriosis-associated cancers. Int J Mol Sci 2024; 25 (14): 7624. doi: 10.3390/ijm s25147624.
155) Suda K, Cruz Diaz LA, Yoshihara K, et al. Clonal lineage from normal endometrium to ovarian clear cell carcinoma through ovarian endometriosis. Cancer Sci 2020; 111 (8): 3000-9. doi: 10.1111/c as.14507.
156) Mutter GL, Monte NM, Neuberg D, Ferenczy A, Eng C. Emergence, involution, and progression to carcinoma of mutant clones in normal endometrial tissues. Cancer Res 2014; 74 (10): 2796-802. doi: 10.1158/0008-5472.CAN-14-0108.
157) Maruyama TA. Revised stem cell theory for the pathogenesis of endometriosis. J Pers Med 2022; 12 (2): 216. doi: 10.3390/jpm12020216.
158) Noë M, Ayhan A, Wang TL, Shih IM. Independent development of endometrial epithelium and stroma within the same endometriosis. J Pathol 2018; 245 (3): 265-9. doi: 10.1002/path.5082.
159) Wilczynski JR, Szubert , Paradowska E, Wilczyński M. Endometriosis stem cells as a possible main target for carcinogenesis of endometriosis-associated ovarian cancer (EAOC). Cancers (Basel) 2022; 15 (1): 111. doi: 10.3390/can cers15010111.
160) Suda K, Nakaoka H, Yoshihara K, et al. Different mutation profiles between epithelium and stroma in endometriosis and normal endometrium. Hum Reprod 2019; 34 (10): 1899-905. doi: 10.109 3/humrep/dez155.
161) Bulun SE, Wang Y, Matei D. Epithelial mutations in endometriosis: link to ovarian cancer. Endocrinology 2019; 160 (3): 626-38. doi: 10.121 0/en.2018-00794.
162) Paik DY, Jansen DM, Schafenacker AM, et al. Stem-like epithelial cells are concentrated in the distal end of the fallopian tube: a site for injury and serous cancer initiation. Stem Cells 2012; 30 (11): 2487-97. doi: 10.1002/stem.1207.
163) Okamoto S, Okamoto A, Nikaido T, et al. Mesenchymal to epithelial transition in the human ovarian surface epithelium focusing on inclusion cysts. Oncol Rep 2009; 21 (5): 1209-14. doi: 10.38 92/or_00000343.
164) Konrad L, Dietze R, Riaz MA, et al. Epithelial-Mesenchymal transition in endometriosis – When does it happen? J Clin Med 2020; 9 (6): 1915. doi: 10.3390/jcm9061915.
165) Ahmed N, Thompson EW, Quinn MA. Epithelial-mesenchymal interconversions in normal ovarian surface epithelium and ovarian carcinomas: an exception to the norms. J Cell Physiol 2007; 213 (3): 581-8. doi: 10.1002/jcp.21240.
166) Okamura H, Katabuchi H. Detailed morphology of human surface epithelium focusing on its metaplastic and neoplastic capability. Ital J Anat Embryol 2001; 106 (2 suppl 2): 263-76.
167) Sugiyama T, Kamura T, Kigawa J, et al. Clinical characteristics of clear cell carcinoma of the ovary: a distinct histologic type with poor prognosis and resistance to platinum-based chemotherapy. Cancer 2000; 88 (11): 2584-9.
168) Schwartz DR, Kardia SL, Shedden KA, et al. Gene expression in ovarian cancer reflects both morphology and biological behavior, distinguishing clear cell from other poor-prognosis ovarian carcinomas. Cancer Res 2002; 62 (16): 4722-9.
169) Yamaguchi K, Huang Z, Matsumura N, et al. Epigenetic determinants of ovarian clear cell carcinoma biology. Int J Cancer 2014; 135 (3): 585-97. doi: 10.1002/ijc.28701.
170) Tsuchiya A, Sakamoto N, Yasuda J, et al. Expression profiling in ovarian clear cell carcinoma: identification of hepatocyte nuclear factor -1 beta as a molecular marker and a possible molecular target for therapy of ovarian clear cell carcinoma. Am J Pathol 2003; 163 (6): 2503-12. doi: 10.10 16/s0002-9440(10)63605-x.
171) Kobayashi H, Yamada Y, Kanayama S, et al. The role of hepatocyte nuclear factor-1 in the pathogenesis of clear cell carcinoma of the ovary. Int J Gynecol Cancer 2009; 19 (3): 471-9. doi: 10.1 111/IGC.0b013e3181a19eca.
172) Kajihara H, Yamada, Shigetomi H, Higashiura Y, Kobayashi H. The dichotomy in the histogenesis of endometriosis-associated ovarian cancer: clear cell-type versus endometrioid-type adenocarcinoma. Int J Gynecol Pathol 2012; 31(4): 304-12. doi: 10.1097/PGP.0b013e318243a97b.
173) Okamoto T, Mandai M, Matsumura N, et al. Hepatocyte nuclear factor-1 (HNF-1) promotes glucose uptake and glycolytic activity in ovarian clear cell carcinoma. Mol Carcinog 2015; 54 (1): 35-49. doi: 10.1002/mc.22072.
174) Mandai M, Amano Y, Yamaguchi K, Matsumura N, Baba T, Konishi I. Ovarian clear cell carcinoma meets metabolism; HNF-1 confers survival benefits through the Warburg effect and ROS reduction. Oncotarget 2015; 6 (31): 30704-14. doi: 10.18632/oncotarget.5228.
175) Kajihara H, Yamada Y, Kanayama S, et al. Clear cell carcinoma of the ovary: Potential pathogenic mechanisms (Review). Oncol Rep 2010; 23 (5): 1193-203. doi: 10.3892/or_00000750.
176) Fujimura M, Hidaka T, Kataoka K, et al. Absence of estrogen receptor-alpha expression in human ovarian clear cell adenocarcinoma compared with ovarian serous, endometrioid, and mucinous adenocarcinoma. Am J Surg Pathol 2001; 25 (5): 667-72. doi: 10.1097/00000478-200105000-00016.
177) Akahane T, Sekizawa A, Okuda T, Kushima M, Saito H, Okai T. Disappearance of steroid hormone dependency during malignant transformation of ovarian clear cell cancer. Int J Gynecol Pathol 2005; 24 (4): 369-76. doi: 10.1097/01.pgp.00001 65173.90339.a2.
178) Yano M, Katoh H, Miyasawa M, et al. Clinicopathological correlation of ARID1A status with HDAC6 and its related factors in ovarian clear cell carcinoma. Sci Rep 2019; 9 (1): 2397. doi: 10.1038/s41598-019-38653-0.
179) Tong A, Xiangjie D, Zhao X, Liang X. Review the progression of ovarian clear cell carcinoma from the perspective of genomics and epigenomics. Front Genet 2023; 14: 952379. doi: 10.3389/fge ne.2023.952379.
180) Mandai M, Matsumura N, Baba T, Yamaguchi K, Hamanishi J, Konishi I. Ovarian clear cell carcinoma as a stress-responsive cancer: Influence of the microenvironment on the carcinogenesis and cancer phenotype. Cancer Lett 2011; 310 (2): 129-33. doi: 10.1016/j.canlet.2011.06.039.
181) Lim D, Murali R, Murray MP, Veras E, Park KJ, Soslow RA. Morphological and immunohistochemical re-evaluation of tumors initially diagnosed as ovarian endometrioid carcinoma with emphasis on high-grade tumors. Am J Surg Pathol 2016; 40 (3): 302-12. doi: 10.1097/PAS.0000000000000550.
182) Hollis RL, Thompson JP, Stanley B, et al. Molecular stratification of endometrioid ovarian carcinoma predicts clinical outcome. Nat Commun 2020; 11 (1): 4995. doi: 10.1038/s41467-020-18819-5.
183) Cochrane DR, Tessier-Cloutier B, Lawrence KM, et al. Clear cell and endometrioid carcinomas: are their differences attributable to distinct cells of origin. J Pathol 2017; 243 (1): 26-36. doi: 10.10 02/path.4934.
184) Kolin DL, Dinulescu DM, Crum CP. Origin of clear carcinoma: nature or nurture? J Pathol 2018; 244 (2): 131-4. doi: 10.1002/path.5009.
185) Beddows I, Fan H, Heinze K, et al. Cell state of origin impacts development of distinct endometriosis-related ovarian carcinoma histotypes. Cancer Res 2024; 84 (1): 26-38. doi: 10.1158/0008-5472.CAN-23-1362.