

Epigenetic Effects of Temozolomide in Glioma Therapy
Epigenetic Mechanisms Underlying the Therapeutic Effects of Temozolomide in Glioma
Tieli Wang1
- Department of Chemistry and Biochemistry, 2Department of Clinical Science, California State University Dominguez Hills, Carson, CA 90747
OPEN ACCESS
PUBLISHED: 31 October 2025
CITATION: Wang, T., 2025. Epigenetic Modulation of Histone Methylation by Temozolomide in Glioma. Medical Research Archives, [online] 13(10). https://doi.org/10.18103/mra.v13i10.6955
COPYRIGHT: © 2025 European Society of Medicine. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
DOI https://doi.org/10.18103/mra.v13i10.6955
ISSN 2375-1924
ABSTRACT
Temozolomide remains the standard chemotherapeutic for glioblastoma multiforme, the most aggressive primary brain tumor. Traditionally, temozolomide has long been understood as a DNA alkylating agent that triggers cell death through O6-methylguanine formation and subsequent mismatch repair-mediated apoptosis. However, emerging research has uncovered that temozolomide also functions as a dual epigenetic regulator. Beyond DNA methylation, temozolomide can modulate histone methylation patterns both through direct chemical modifications and indirect effects on histone-modifying enzyme activity. These mechanisms fundamentally reorganize chromatin architectures and alter gene expression programs within cancer cells. The combined genotoxic and epigenetic actions of temozolomide not only contribute to its anti-tumor effectiveness but also drive adaptive mechanisms of cancer cell resistance and recurrence. Understanding temozolomide’s capacity for epigenetic reprogramming and its impact on tumor microenvironment dynamics provides crucial insights for developing more effective treatment strategies. This knowledge supports the rationale for combination therapies that integrate temozolomide with epigenetic inhibitors, targeted enzyme modulators, or bioactive natural compounds alongside radiation therapy, potentially leading to significantly improved clinical outcomes for glioma patients.
Keywords: Temozolomide, therapeutic benefit, DNA and histone methylation, Cell death pathways, epigenetic regulation
Introduction
Glioblastoma multiforme (GBM) is the most aggressive and common primary malignant tumor of the central nervous system, characterized by rapid progression, extensive infiltration into surrounding brain tissue, and profound therapeutic resistance. Despite advances in multimodal treatment, prognosis remains dismal, with a median survival of only approximately 15 months and a five-year survival rate below 10%. The current standard of care involves maximal surgical resection followed by radiotherapy and in combination with concomitant and adjuvant chemotherapy using temozolomide (TMZ). This regimen has significantly improved overall survival compared to radiotherapy alone, establishing TMZ as a standard chemotherapeutic agent of systemic therapy in GBM. As the second-generation oral alkylating agent, TMZ remains the frontline chemotherapeutic drug for gliomas due to its ability to cross the blood-brain barrier and induce DNA alkylation, primarily at the O6-position of guanine, leading to cytotoxic DNA damage and apoptotic cell death.
Although TMZ has provided measurable survival benefits both as monotherapy and in combination with radiotherapy and surgery, its long-term clinical efficacy remains severely limited. The principal challenge lies in the intratumoral heterogeneity and the intrinsic and acquired adaptive capacity of glioma cells. Tumor heterogeneity, dynamic interactions with the tumor microenvironment and molecular plasticity allow malignant cells to rapidly develop resistance mechanisms, including DNA repair, activation of compensatory signaling pathways, and epigenetic reprogramming. As a result, recurrence is almost universal, and durable responses to TMZ are rare.
In this review, the focus of the discussion will be specifically on the intersection of epigenetic regulation and temozolomide therapy in glioma. Increasing evidence suggests that TMZ not only exerts genotoxic stress but also induces widespread epigenetic alterations, including dynamic changes in DNA and histone methylation, which in turn reshape gene expression programs. These modifications may act as double-edged swords promoting tumor suppression in some contexts while facilitating tumor adaptation and progression in others. Understanding these epigenetic consequences of TMZ exposure is therefore critical for developing rational combinatorial strategies with radiotherapy. Potential approaches to enhance the therapeutic impact of TMZ include (i) targeting epigenetic mechanisms in glioma, for example by combining TMZ with inhibitors of chromatin-modifying enzymes to prevent adaptive transcriptional reprogramming; (ii) exploring natural products as alternatives to TMZ and developing natural product-based chemoradiotherapy regimens; (iii) employing nanoparticle-based or ligand-directed delivery systems to improve drug specificity and overcome the limitations of systemic administration. While the broader discussion of glioma treatment resistance and therapeutic failures will be addressed in a separate publication, the present review aims to provide mechanistic insights into how epigenetic regulation shapes the therapeutic potential of temozolomide in glioma. By integrating knowledge of DNA and protein damage responses with epigenetic plasticity, we may uncover new opportunities for precision therapies that extend survival and improve clinical outcomes for patients with glioma.
a. PROTEIN-LEVEL EFFECTS AND HISTONE METHYLATION BY TEMOZOLOMIDE
TMZ works by spontaneously producing a methylating metabolite under physiological conditions through a chemical process:
- TMZ undergoes rapid chemical conversion in the bloodstream at physiological pH.
- It produces a metabolite called 5-(3-methyltriazen-1-yl) imidazole-4-carboxamide (MTIC).
- MTIC further degrades into two components as shown in Figure 1:
- 4-amino-5-imidazole-carboxamide (AIC): An inactive metabolite
- Methyldizaonium cation: The active methylating agent
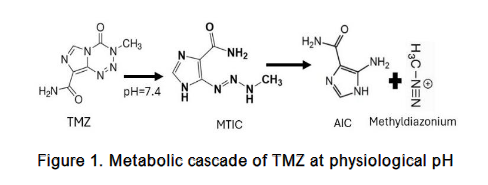
The antitumor effect of TMZ through DNA methylation is well established. This DNA methylation damages DNA and induces cancer cell death. However, the antitumor activity of TMZ on proteins remains poorly understood. Proteins, as the primary executors of cellular activity, play diverse and crucial roles in living organisms, surpassing genes in both numbers and functional complexity. Protein methylation is a type of post-translational modification which involves chemical modifications of amino acid side chains, particularly on lysine and arginine residues. Histones are now recognized as dynamic proteins that undergo multiple types of post-translational modifications. The importance of histone methylation lies in its crucial role in determining active and inactive regions of the genome, making it highly significant for cancer and other disease studies. We believe that the therapeutic benefits of temozolomide also extend to protein levels, especially affecting histones and their associated proteins. Histones provide structural support for chromosomes. Each chromosome contains a long molecule of DNA that must be wrapped around histones to fit into a small space in the nucleus of cells, giving the chromosomes their compact structure. Therefore, alterations in histone structure can significantly influence gene expression and genomic stability. Our in vitro biochemical assay using histone H3 peptides and full-length histone I proteins revealed TMZ methylates specific lysine and arginine residues through a non-enzymatic mechanism. The histone methylation levels exhibit an inverse relationship with the temozolomide dosage in U87 glioma cells. These post-translational modifications were validated via mass spectrometry and Western blotting, demonstrating TMZ’s ability to directly modify histone architecture independently of conventional histone methyltransferases. Furthermore, TMZ affected the expression and activity of histone lysine demethylases, indicating a dual mode of action: direct methylation of histone residues and indirect regulation of histone methylation dynamics. This dual functionality positions TMZ as a multi-faceted epigenetic modulator capable of reshaping chromatin structure and gene expression. Such epigenetic alterations may synergize with DNA damage responses, enhancing TMZ’s therapeutic efficacy through both cytotoxic and transcriptional mechanisms. Investigating the impact of temozolomide on histone methylation levels could provide valuable insights into its potential therapeutic benefits at the protein levels.
b. SITE-SPECIFIC EFFECTS OF HISTONE METHYLATION
At present, the only study investigating the effects of TMZ on site-specific histone methylation has identified changes at histone H3 lysine 4 (H3K4) residues. Beyond this, the literature on TMZ and histone methylation remains limited. Therefore, this section will consider site-specific alterations in histone methylation reported in other contexts, particularly those involving histone-modifying enzymes, while acknowledging that such mechanisms may, or may not be applicable to TMZ.
Methylations of H3K4 are among the most extensively studied histone modifications due to their association with histone methylating enzymes and gene expression in cancers. This includes mono-, di- and tri-methylation and some of the best characterized substrates are histone H3 lysine 9 (H3K9), lysine 27 (H3K27), lysine 36 (H3K36) and lysine 79 (H3K79) and histone H4 lysine 20 (H4K20). These methyl marks can contribute to regulation of transcription frequently acting as leading platforms for the recruitment of effector proteins. Histone lysine methylation is also associated with other diverse functions including heterochromatin formation, X chromosome inactivation, DNA repair, cell fate determination and terminal differentiation. Dysregulation of histone lysine methylation is associated with several human cancers and other diseases.
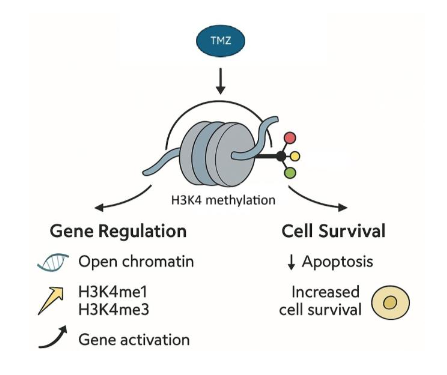
H3K4 Methylation: A Critical Survival Signal
H3K4 methylation has site-specific effects that can activate or repress genes by altering chromatin structure and recruiting or blocking binding proteins. Different lysine residues on the histone H3 tail, and the degrees of their methylation (mono-, di-, or tri), dictate the functional outcomes. Methylation at H3K4, especially H3K4me1 and me3 may have convergent functions in establishing open chromatin and are generally considered to be associated with gene activation. H3K4me3 variation is mostly correlated with the level of transcription. H3K4me2 shares features with both H3K4me1 and H3K4me3 and probably represents an intermediate or transitional state. H3K4me1 functions to activate enhancer regions involved in gene transcription.
H3K4 methylation plays a particularly crucial role in cell survival and apoptosis regulation. In yeast models, loss of H3K4 methylation correlates with increased cell death during aging and enhanced apoptotic sensitivity. In mouse model, H3K4 mutation is associated with neuronal differentiation defects. This relationship is further supported by observations that aged and dying wild-type yeast cells naturally lose H3K4 methylation marks. Importantly, depletion of H3K4 demethylases- the enzymes responsible for removing these marks improves cellular survival, confirming the protective role of this modification.
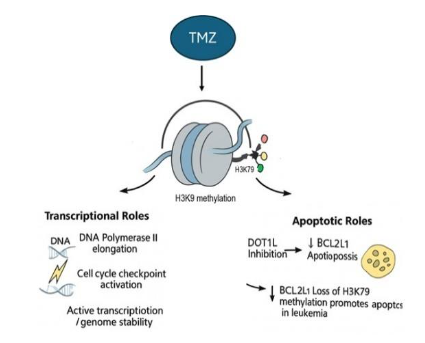
H3K79 Methylation: Context-Dependent Effects
Unlike H3K9 and H3K27 methylation, which are predominantly associated with gene silencing, H3K79 Methylation plays a role in active transcriptional processes, including transcription elongation by RNA polymerase II, DNA damage response and cell cycle checkpoint regulation. Uniquely among histone lysine modifications, H3K79 methylation currently lacks an identified histone demethylase. However, there is considerable evidence suggesting that H3K79 methylation is reversible.
Histone demethylases function within larger multiprotein complexes alongside histone deacetylases, histone methyltransferases and nuclear receptors to regulate developmental and transcriptional programs. Rapid depletion of H3K79me2 has been observed during early development in both flies and mice. In addition, factors affecting the rate of cell division and replication independent histone turnover affect both the level of H3K79 methylation and its distribution across the genomic. H3K79me2 serves as a marker of actively transcribed genes.
The functional outcomes of H3K79 methylation vary based on the degree of methylation at this residue. Research indicates that removal of H3K79 methylation following fertilization contributes to epigenetic reprogramming. H3K79 methylation typically exerts more subtle effects compared to H3K4 methylation in yeast systems, nonetheless it still plays important roles in regulating cell death under specific conditions. In certain leukemia cell lines, inhibition of the H3K79 methyltransferase induces apoptosis, likely by suppressing expression of the anti-apoptotic protein BCL2L1.
c. CELL DEATH PATHWAYS
Beyond classical apoptosis, histone methylation has emerged as a critical regulator of alternative forms of programmed cell, including autophagy and ferroptosis. For instance, H3K4 methylation catalyzed by methyltransferases such as SETD1A and MLL family proteins, is generally linked to transcriptional activation and can promote the expression of autophagy-related genes. In contrast, repressive marks like H3K9me2/3, deposited by SUV39H1 or G9A, and H3K27me3, mediated by EZH2 within the Polycomb repressive complex 2 (PRC2), can silence genes involved in autophagy or stress responses, thereby restraining survival pathways. Demethylases also play important roles; for example, KDM5 family proteins remove H3K4 methylation to repress autophagy, while KDM6A/B demethylate H3K27me3 to depress stress-induced gene programs. In the context of ferroptosis, histone methylation has been shown to influence transcription of genes regulating iron metabolism (e.g., transferring receptor), lipid peroxidation, and antioxidant defenses such as GPX4, thereby shaping cellular susceptibility to oxidative damage. The interplay of these enzymes and their respective histone marks underscores the capacity of histone methylation to either promote or restrain distinct death pathways, highlighting its fundamental role in maintaining cellular homeostasis. Collectively, methylation at H3K4, H3K9 and H3K27 extends well beyond apoptosis to encompass diverse nuclear processes, including transcriptional activation, repression and autophagy. This multifunctionality underscores how epigenetic modifications serve as central integrators of cellular signaling networks.
Additionally, histone methylation contributes to the structural reorganization of chromatin during apoptosis, facilitating the characteristic nuclear changes that accompany programmed cell death. This structural role complements the regulatory functions of these modifications in controlling gene expression programs that determine cell fate. Histone methylation is crucial in regulating apoptosis by modulating gene expression that either induces or prevents programmed cell death. The therapeutic potential of reversing these epigenetic alterations has shown efficacy in lymphomas and pre-leukemic disorders, with encouraging outcomes and now extending to solid tumors. Emerging evidence reveals that combinations of histone modifications, rather than individual marks, influence cellular processes including programmed cell death, emphasizing the complex regulatory landscape of histone methylation in cell fate decisions.
d. TMZ-MEDIATED EPIGENETIC REPROGRAMMING VIA P53
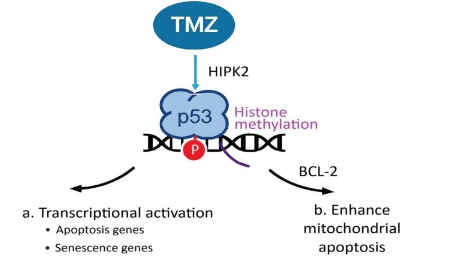
The Cancer Genome Atlas (TCGA) data reveals that p53 mutations occur in 31% of glioblastomas and 48% of low-grade gliomas. P53 Status in Gliomas and Its Functional Role have been studied. As a critical transcription factor, p53 orchestrates the expression of genes maintaining genomic stability, facilitating chromatin remodeling, governing cell cycle regulation, DNA repair, apoptosis, and numerous other cellular processes. Functioning as a tumor suppressor, p53 promotes cell cycle arrest or triggers apoptosis in response to DNA damage or cellular stress.
TMZ may or may not methylate P53 directly to affect its activity, instead it serves as potent p53 activators. Study has shown that lysine methylation regulates p53 activity. The TMZ-p53 relationship is fundamental to gene expression regulation, genomic stability maintenance, chromatin remodeling facilitation, and damage repair promotion. The interplay between TMZ and p53 involves multiple interconnected mechanisms that can operate simultaneously.
DIRECT TRANSCRIPTIONAL REGULATION
TMZ activates P53 by phosphorylation of P53 at Ser46 through SIAH1-HIPK2 kinase pathways and directly regulates numerous pro-apoptotic target genes, including FAS, BAX, BAK, PTEN, and PUMA. Additionally, p53 can interact with BCL2 family proteins and enhance mitochondrial apoptosis through mitochondrial membrane translocation and cytochrome C release. TMZ plays important role in promoting transcriptional activation and enhancing mitochondrial apoptosis through P53 activation. These interactions facilitate the recruitment of regulatory complexes to target genes, where histone modifications near p53 binding sites can either enhance or suppress gene expression.
CHROMATIN-MEDIATED EFFECTS EPIGENETIC REGULATION
In addition to DNA methylation, TMZ influences the activity and expression of histone-modifying enzymes, including histone methyltransferases and demethylases such as enhancer of Zeste Homolog 2 (EZH2), various lysine demethylases (KDMs) and transcription factors like P53 and NFkB. These enzymes regulate histone methylation marks associated with chromatin remodeling, gene expression, and damage response pathways. Under TMZ treatment conditions, altered expression of these enzymes may drive epigenetic reprogramming, contributing to either therapeutic sensitivity or acquired resistance in glioblastoma cells.
Although direct studies investigating the chromatin-level interplay between TMZ and p53 are currently lacking, accumulating mechanistic insights suggest that TMZ may exert indirect effects on p53 activity through modulation of chromatin architecture. TMZ-mediated histone methylation can trigger alterations in chromatin structure, thereby influencing the accessibility of transcription factor binding sites, including those recognized by p53.
Specifically, TMZ may shift chromatin between euchromatic (open) and heterochromatic (closed) states by modulating histone modifications such as methylation and acetylation. These changes can significantly affect the ability of p53 to bind the promoters or enhancers of its target genes, thereby influencing the transcriptional regulation of genes involved in apoptosis, cell cycle and cell state control. Conversely, p53 itself can respond to TMZ-induced cellular stress by directly regulating the transcription of genes encoding histone-modifying enzymes and chromatin-associated proteins. For instance, p53 binding at the promoters of such genes could influence histone methyltransferases, demethylases, acetyltransferases, or chromatin remodeling complexes, thereby establishing a feedback loop in which TMZ-induced epigenetic changes and p53 activity are mutually reinforcing.
The possibility of bidirectional relationship implies that the functional outcome of TMZ treatment is not solely determined by direct DNA and protein damage, but also by epigenetic reprogramming events that fine-tune p53-mediated transcriptional responses. Elucidating these dynamic mechanisms could uncover novel regulatory layers contributing to glioma response to TMZ therapy and may point to epigenetic targets for combination strategies. This gap in knowledge represents a critical opportunity to advance the field of cancer epigenetics. A more comprehensive understanding of TMZ’s epigenetic effects could lead to the development of combination therapies that enhance its efficacy or overcome resistance. Specifically, targeting the histone modification landscape in concert with DNA methylation may provide a more robust strategy for treating glioblastoma and improving patient outcomes.
e. EPIGENETIC ORCHESTRATION IN GLIOMA
TMZ exerts a dual impact on DNA and histone methylation, positioning it as a central driver of epigenetic reprogramming in glioma. Chromatin accessibility and higher order structure are governed by a dynamically and reversibly interplay of epigenetic machinery. This regulatory system is composed of three main classes of protein:
- Writers, such as DNA methyltransferases and histone methyltransferases (HMTs), catalyze the addition of methyl groups to DNA or histone residues. Members of the SET domain family, for instance, add methyl marks to specific histone lysine and arginine residues, thereby influencing chromatin condensation and transcriptional outcomes.
- Readers, including bromodomain-containing proteins and other recognition factors, interpret these specific epigenetic marks and recruit co-regulatory complexes that enforce transcriptional states.
- Erasers, such as lysine-specific demethylase 1 (LSD1) and Jumonji C (JMJC) domain containing demethylases, remove methyl groups to re-establish transcriptional flexibility.
Together, these components orchestrate chromatin dynamics and transcriptional programs. Unlike other post-translational modifications (e.g. acetylation or phosphorylation), histone methylation doesn’t alter substrate charges. Instead, its functional consequence arises primarily through the recruitment of specialized reader proteins that recognize distinct methylation patterns.
Dysregulation of these enzymes and their pathways can profoundly influence glioma cell responses to TMZ. Thus, dissecting the molecular mechanisms by which TMZ reshapes the epigenetic landscape is essential for clarifying its mode of action and identifying new therapeutic opportunities. While many features of glioma epigenomes remain incompletely understood, accumulating evidence indicates that DNA and histone methylation define key regulatory elements and shape therapeutic response of TMZ.
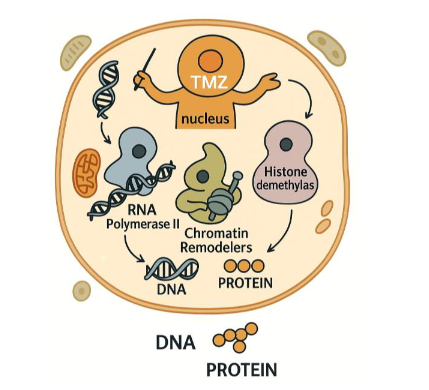
More broadly, glioma epigenomes encompass a complex landscape of DNA methylation, histone modifications, and non-coding RNA regulation. These epigenetic layers collectively shape gene expression programs and post translational signaling networks, influencing cellular processes such as metabolism, redox balance, autophagy, proliferation and apoptosis. While many features of glioma epigenomes remain incompletely understood, accumulating evidence indicates that DNA and histone methylation define key regulatory elements and shape therapeutic response of TMZ.
Emerging therapeutic strategies are exploiting these vulnerabilities. For example, epigenetic modulation of gene transcription using a quantitative systems pharmacology (QSP) model of TMZ introduces a new therapeutic concept, Cell State-Directed (CSD) therapy, for overcoming TMZ resistance in glioma. This strategy targets dynamic transitions between cancer cells states through epigenetic modulation of gene transcription, disrupting tumor survival microenvironment and mechanisms. Targeting these cell state transitions can enhance TMZ’s effectiveness.
Natural products also represent an intriguing frontier. Compounds that mimic the alkylating activity of TMZ while potentially offering improved efficacy and reduced toxicity, may provide valuable alternatives or adjuncts to existing regimens. Their integration with radiotherapy protocols could further amplify therapeutic benefits.
Collectively, these insights highlight the multi-faceted role of epigenetic regulation in glioma and underscore the potential of rationally designed combination therapies. A deeper understanding of TMZ-induced epigenetic remodeling not only clarifies its mechanism of action but also opens avenues for the development of novel epigenetic agents and integrative treatment strategies to improve outcomes in glioma.
Conclusion
The therapeutic benefit of TMZ extends beyond its DNA-alkylating activity. Its ability to methylate proteins, especially histones and histone-associated enzymes in glioma, demonstrates its broader biological effects. TMZ functions not merely as a genotoxic agent but as an epigenetic modulator that influences chromatin states, transcriptional programs, and tumor adaptability. This dual mechanism of action—combining direct DNA and protein methylation with epigenetic regulation—enables TMZ to reprogram tumor cell states, highlighting its multifaceted role in cancer therapy.
Further exploration of these underlying molecular processes will enhance our comprehension of TMZ-induced epigenetic alterations, and how they interface with genetic, metabolic and microenvironmental factors. It will be essential for the rational design of novel combination therapies. Such research holds promise for developing more effective treatment approaches that could significantly improve clinical outcomes for patients facing this particularly challenging malignancy.
Conflict of interests: The authors declare that there is no conflict of interest.
Acknowledgements: The author gratefully acknowledges the sabbatical leave granted by California State University Dominguez Hills, which enabled the investigation of histone methylation induced by temozolomide. Special thanks are extended to Dr. James Gallo of Icahn School of Medicine at Mount Sinai, New York, for his generous invitation to join his laboratory as a visiting professor and for his invaluable guidance throughout the visit. The author appreciates the assistance and discussions from ChatGPT, made available through California State University, during the document editing process.
References:
- Koukourakis GV, Kouloulias V, Zacharias G, et al. Temozolomide with radiation therapy in high grade brain gliomas: pharmaceuticals considerations and efficacy; a review article. Molecules. 2009;14(4):1561-1577. Published 2009 Apr 16. doi:10.3390/molecules14041561
- Schaff LR, Mellinghoff IK. Glioblastoma and Other Primary Brain Malignancies in Adults: A Review. JAMA. 2023;329(7):574-587. doi:10.1001/jama.2023.0023
- Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987-996. doi:10.1056/NEJMoa043330
- Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459-466. doi:10.1016/S1470-2045(09)70025-7
- Kaina B. Temozolomide, Procarbazine and Nitrosoureas in the Therapy of Malignant Gliomas: Update of Mechanisms, Drug Resistance and Therapeutic Implications. J Clin Med. 2023;12(23):7442. Published 2023 Nov 30. doi:10.3390/jcm12237442
- Roos WP, Kaina B. DNA damage-induced cell death: from specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013;332(2):237-248. doi:10.1016/j.canlet.2012.01.007
- Fares J, Petrosyan E, Dmello C, Lukas RV, Stupp R, Lesniak MS. Rethinking metastatic brain cancer as a CNS disease. Lancet Oncol. 2025;26(2):e111-e121. doi:10.1016/S1470-2045(24)00430-3
- Caldwell BA, Bartolomei MS. DNA methylation reprogramming of genomic imprints in the mammalian germline: A TET-centric view. Andrology. 2023;11(5):884-890. doi:10.1111/andr.13303
- Jezierzański M, Nafalska N, Stopyra M, et al. Temozolomide (TMZ) in the Treatment of Glioblastoma Multiforme-A Literature Review and Clinical Outcomes. Curr Oncol. 2024;31(7):3994-4002. Published 2024 Jul 12 doi:10.3390/curroncol31070296
- Danson SJ, Middleton MR. Temozolomide: a novel oral alkylating agent. Expert Rev Anticancer Ther. 2001;1(1):13-19. doi:10.1586/14737140.1.1.13
- Barczewski AM, Gurda D, Godowicz P, Nowak S, Naskręt-Barciszewska MZ. A New Epigenetic Mechanism of Temozolomide Action in Glioma Cells. PLoS One. 2015;10(8):e0136669. Published 2015 Aug 26. doi:10.1371/journal.pone.0136669
- Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31(2):89-97. doi:10.1016/j.tibs.2005.12.008
- Chuikov S, Kurash JK, Wilson JR, et al. Regulation of p53 activity through lysine methylation. Nature. 2004 Nov 18;432(7015):353-60. doi:10.1038/nature03117. Epub 2004 Nov 3. PMID: 15525938.
- England B, Huang T, Karsy M. Current understanding of the role and targeting of tumor suppressor p53 in glioblastoma multiforme. Tumour Biol. 2013;34(4):2063-2074. doi:10.1007/s13277-013-0871-3
- He Y, Kaina B. Are There Thresholds in Glioblastoma Cell Death Responses Triggered by Temozolomide? Int J Mol Sci. 2019;20(7):1562. Published 2019 Mar 28.doi:10.3390/ijms20071562
- Aasland D, Götzinger L, Hauck L, et al. Temozolomide Induces Senescence and Repression of DNA Repair Pathways in Glioblastoma Cells via Activation of ATR-CHK1, p21, and NF-κB. Cancer Res. 2019;79(1):99-113. doi:10.1158/0008-5472.CAN-18-1733
- Tomicic MT, Meise R, Aasland D, et al. Apoptosis induced by temozolomide and nimustine in glioblastoma cells is supported by JNK/c-Jun-mediated induction of the BH3-only protein BIM. Oncotarget. 2015;6(32):33755-33768. doi:10.18632/oncotarget.5274
- Avci NG, Ebrahimzadeh-Pustchi S, Akay YM, et al. NF-κB inhibitor with Temozolomide results in significant apoptosis in glioblastoma via the NF-κB(p65) and actin cytoskeleton regulatory pathways. Sci Rep. 2020;10(1):13352. Published 2020 Aug 7 doi:10.1038/s41598-020-70392-5
- Kaina B. Temozolomide, Procarbazine and Nitrosoureas in the Therapy of Malignant Gliomas: Update of Mechanisms, Drug Resistance and Therapeutic Implications. J Clin Med. 2023;12(23):7442. Published 2023 Nov 30. doi:10.3390/jcm12237442
- Caporali S, Falcinelli S, Starace G, et al. DNA damage induced by temozolomide signals to both ATM and ATR: role of the mismatch repair system. Mol Pharmacol. 2004;66(3):478-491. doi:10.1124/mol.66.3.
- Pickard J., Diaz AJ, Mura, H., Nyuwen L, Coello, D., Saif, S., Nava, M., Gallo, J., and Wang T. Histone Methylation by Temozolomide; A Classic DNA Methylating Anticancer Drug. Supplements: World Biomedical frontiers, 2016.
- Wang T, Pickard AJ, Gallo JM. Histone Methylation by Temozolomide; A Classic DNA Methylating Anticancer Drug. Anticancer Res. 2016;36(7):3289-3299.
- Martinez, P, Bernal, R, Hills, R, and Wang, T. Anti-cancer drug, temozolomide, increased histone demethylase activity in breast cancer cells. 2024 Annual symposium of CSUPERB, Santa Clare, CA.
- Füllgrabe J, Hajji N, Joseph B. Cracking the death code: apoptosis-related histone modifications. Cell Death Differ. 2010;17(8):1238-1243. doi:10.1038/cdd.2010.58
- Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell. 2007;25(1):15-30. doi:10.1016/j.molcel.2006.12.014
- Shilatifard A. Molecular implementation and physiological roles for histone H3 lysine 4 (H3K4) methylation. Curr Opin Cell Biol. 2008;20(3):341-348. doi:10.1016/j.ceb.2008.03.019
- Kouzarides T. Histone methylation in transcriptional control. Curr Opin Genet Dev. 2002;12(2):198-209. doi:10.1016/s0959-437x(02)00287-3
- Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 2005;6(11):838-849. doi:10.1038/nrm1761
- Black JC, Van Rechem C, Whetstine JR. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell. 2012;48(4):491-507. doi:10.1016/j.molcel.2012.11.006
- Handel AE, Ebers GC, Ramagopalan SV. Epigenetics: molecular mechanisms and implications for disease. Trends Mol Med. 2010;16(1):7-16. doi:10.1016/j.molmed.2009.11.003
- Rajan PK, Udoh UA, Sanabria JD, et al. The Role of Histone Acetylation-/Methylation-Mediated Apoptotic Gene Regulation in Hepatocellular Carcinoma. Int J Mol Sci. 2020;21(23):8894. Published 2020 Nov 24. doi:10.3390/ijms21238894
- Yu H, Lesch BJ. Functional Roles of H3K4 Methylation in Transcriptional Regulation. Mol Cell Biol. 2024;44(11):505-515. doi:10.1080/10985549.2024.2388254
- Wang H, Helin K. Roles of H3K4 methylation in biology and disease. Trends Cell Biol. 2025;35(2):115-128. doi:10.1016/j.tcb.2024.06.001
- Heintzman ND, Hon GC, Hawkins RD, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459(7243):108-112. doi:10.1038/nature07829
- Herz HM, Mohan M, Garruss AS, et al. Enhancer-associated H3K4 monomethylation by Trithorax-related, the Drosophila homolog of mammalian Mll3/Mll4. Genes Dev. 2012;26(23):2604-2620. doi:10.1101/gad.201327.112
- Briggs SD, Strahl BD. Unraveling heterochromatin. Nat Genet. 2002 Mar;30(3):241-2. doi:10.1038/ng0302-241. PMID: 11919553.
- Fahrenkrog B. Histone modifications as regulators of life and death in Saccharomyces cerevisiae. Microb Cell. 2015 Dec 31;3(1):1-13. doi:10.15698/mic2016.01.472. PMID: 28357312; PMCID: PMC5354586.
- Walter D, Matter A, Fahrenkrog B. Loss of histone H3 methylation at lysine 4 triggers apoptosis in Saccharomyces cerevisiae. PLoS Genet. 2014;10(1):e1004095. Published 2014 Jan 30. doi:10.1371/journal.pgen.1004095
- Gehre M, Bunina D, Sidoli S, et al. Lysine 4 of histone H3.3 is required for embryonic stem cell differentiation, histone enrichment at regulatory regions and transcription accuracy. Nat Genet. 2020;52(3):273-282. doi:10.1038/s41588-020-0586-5
- Wood K, Tellier M, Murphy S. DOT1L and H3K79 Methylation in Transcription and Genomic Stability. Biomolecules. 2018;8(1):11. Published 2018 Feb 27. doi:10.3390/biom8010011
- Caldwell BA, Bartolomei MS. DNA methylation reprogramming of genomic imprints in the mammalian germline: A TET-centric view. Andrology. 2023;11(5):884-890. doi:10.1111/andr.13303
- Cloos PA, Christensen J, Agger K, Helin K. Erasing the methyl mark: histone demethylases at the center of cellular differentiation and disease. Genes Dev. 2008;22(9):1115-1140. doi:10.1101/gad.1652908
- Wysocka J, Milne TA, Allis CD. Taking LSD 1 to a new high. Cell. 2005;122(5):654-658. doi:10.1016/j.cell.2005.08.022
- Shanower GA, Muller M, Blanton JL, et al. Characterization of the grappa gene, the Drosophila histone H3 lysine 79 methyltransferases. Genetics. 2005;169(1):173-184. doi:10.1534/genetics.104.033191
- Ooga M, Inoue A, Kageyama S, et al. Changes in H3K79 methylation during preimplantation development in mice. Biol Reprod. 2008;78(3):413-424. doi:10.1095/biolreprod.107.063453
- Lu Y, Chan YT, Tan HY, et al. Epigenetic regulation in human cancer: the potential role of epi-drug in cancer therapy. Mol Cancer. 2020;19(1):79. Published 2020 Apr 27. doi:10.1186/s12943-020-01197-3
- Hwang YK, Lee DH, Lee EC, Oh JS. Importance of Autophagy Regulation in Glioblastoma with Temozolomide Resistance. Cells. 2024;13(16):1332. doi:10.3390/cells13161332
- Jeon M, Park J, Yang E, Baek HJ, Kim H. Regulation of autophagy by protein methylation and acetylation in cancer. J Cell Physiol. 2022;237(1):13-28. doi:10.1002/jcp.30502
- Lan T, Sun TT, Wei C, et al. Epigenetic Regulation of Ferroptosis in Central Nervous System Diseases. Mol Neurobiol. 2023;60(7):3584-3599. doi:10.1007/s12035-023-03267-1
- Bates SE. Epigenetic Therapies for Cancer. N Engl J Med. 2020;383(7):650-663. doi:10.1056/NEJMra1805035
- Batista LF, Roos WP, Christmann M, et al. Differential sensitivity of malignant glioma cells to methylating and chloroethylating anticancer drugs: p53 determines the switch by regulating xpc, ddb2, and DNA double-strand breaks. Cancer Res. 2007;67(24):11886-11895. doi:10.1158/0008-5472.CAN-07-2964
- England B, Huang T, Karsy M. Current understanding of the role and targeting of tumor suppressor p53 in glioblastoma multiforme. Tumour Biol. 2013;34(4):2063-2074. doi:10.1007/s13277-013-0871-3
- He Y, Kaina B. Are There Thresholds in Glioblastoma Cell Death Responses Triggered by Temozolomide?. Int J Mol Sci. 2019;20(7):1562. Published 2019 Mar 28. doi:10.3390/ijms20071562
- Sun X, Klingbeil O, Lu B, et al. BRD8 maintains glioblastoma by epigenetic reprogramming of the p53 network. Nature. 2023;613(7942):195-202. doi:10.1038/s41586-022-05551-x
- Hafner A, Bulyk ML, Jambhekar A, Lahav G. The multiple mechanisms that regulate p53 activity and cell fate. Nat Rev Mol Cell Biol. 2019;20(4):199-210. doi:10.1038/s41580-019-0110-x
- Bennett RL, Licht JD. Targeting Epigenetics in Cancer. Annu Rev Pharmacol Toxicol. 2018 Jan 6; 58:187-207. doi:10.1146/annurev-pharmtox-010716-105106. Epub 2017 Oct 6. PMID: 28992434; PMCID: PMC5800772.
- Baylin SB, Jones PA. A decade of exploring the cancer epigenome – biological and translational implications. Nat Rev Cancer. 2011;11(10):726-734. Published 2011 Sep 23. doi:10.1038/nrc3130