

Fetal Acidosis and Cerebral Palsy: Mechanisms Explored
How Does Fetal Acidosis Result in Cerebral Palsy?
Michael G. Ross1, MD, MPH
- Distinguished Professor of Obstetrics and Gynecology and Public Health
Geffen School of Medicine at UCLA
Fielding School of Public Health at UCLA
Co-Director Institute for Women’ and Children’s Health
The Lundquist Institute
OPEN ACCESS
PUBLISHED: 28 February 2026
CITATION: Ross, MG., 2026. How Does Fetal Acidosis Result in Cerebral Palsy? Medical Research Archives, [online] 14(2). https://doi.org/10.18103/mra.v14i2.7212
COPYRIGHT: © 2025 European Society of Medicine. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
DOI https://doi.org/10.18103/mra.v14i2.7212
ISSN 2375-1924
ABSTRACT
Cerebral palsy presents enormous challenges to the physical and emotional wellbeing of the affected individual as well as the immediate family. The average lifetime cost per person with cerebral palsy exceeds one million dollars in the United States. While highly dependent on the degree of severity, children severely impaired in cognition, motor skills, hearing, and vision have a markedly shortened life span. Whereas hypoxia-ischemia is considered to be etiologic in fetal/newborn cerebral palsy, the cellular mechanisms by which acidosis, hypoxemia and hypercapnia induce neuronal damage are complex. Questions remain as to why most newborns survive intact despite severe metabolic acidosis (base deficit greater than 12 mmol/L) and whether early newborn biochemical correction of metabolic acidosis may reduce long term injury. This review aims to address the putative pathways by which acidosis, hypoxemia and hypercapnia may result in cell damage. A knowledge of these mechanisms is essential for the development of therapeutic interventions for the prevention of cerebral palsy in at risk newborns.
Keywords
Cerebral palsy, fetal acidosis, hypoxemia, hypercapnia, neonatal injury
Introduction
It is well accepted that fetal severe hypoxemia/ischemia is associated with the risk of the development of neonatal encephalopathy and cerebral palsy. Established criteria for attributing neonatal cerebral palsy to an in utero or newborn hypoxic-ischemic event include an umbilical artery or newborn arterial pH less than 7.0 and/or a base deficit greater than or equal to 12 mmol/L. However, the mechanisms by which acidosis, hypoxemia or the often associated hypercapnia cause cell damage and brain dysfunction are poorly delineated. Only a small percentage of infants with umbilical artery base deficit 12 to 20 mmol/L will manifest cerebral palsy, raising the question as to whether this represents an individual tolerance/susceptibility or whether other associated factors contribute importantly to brain injury. Furthermore, infants born with severe metabolic acidosis have minimal acid clearance in the first one to two hours of life, despite often rapid resuscitation. It is unknown whether the chronicity of severe acidosis in the presence of reoxygenation contributes to cellular dysfunction and whether more rapid biochemical correction of acidosis would improve outcome. This review addresses the current understanding of the pathways of acidosis, hypoxemia, and hypercapnia-induced brain damage. These mechanisms represent targets for the development of therapeutic interventions for the prevention of cerebral palsy in at risk newborns.
Hypoxemia and Acidosis
Ultimately, fetal/newborn acidemia is a consequence of hypoxemia, with the exception of select infant inborn errors of metabolism, uncommon renal or gastrointestinal etiologies, sepsis and response to moderate to severe maternal acidemia. An interruption of the maternal-placental-fetal oxygen pathway, such that interruption of maternal oxygenation (e.g., pulmonary function, cardiac output, systemic blood pressure, uterine perfusion, blood oxygen carrying capacity), placental capability and function, or fetal uptake and distribution (e.g., umbilical perfusion, oxygen carrying capacity) may result in fetal hypoxemia. An estimated 70-80% of cerebral palsy cases are estimated to occur prenatally (i.e., congenital cerebral palsy), with the remainder occurring intrapartum or during early life.
Fetal hypoxemia causes acidosis mainly by forcing cells to reduce or halt mitochondrial oxidative phosphorylation, shifting to make ATP via anaerobic glycolysis, which generates excess acid. Specifically, glycolysis converts glucose to pyruvate, which under low oxygen is reduced to lactate, producing hydrogen ions (H⁺) and causing a metabolic (lactic) acidosis.
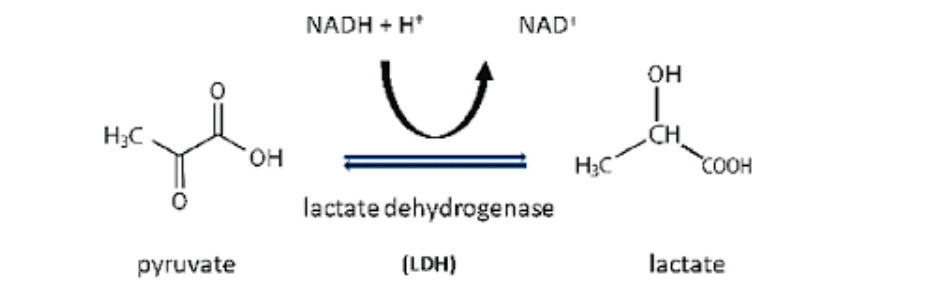
Acidosis itself promotes lactate efflux from within the cells by increasing the activity of specific lactate/H⁺ monocarboxylate cotransporters (MCTs) in the cell membrane, which move lactate out together with protons down their electrochemical gradients. In addition to MCT coupling, sodium-coupled lactate transport occurs. With high levels of intracellular lactate and H⁺, MCT-mediated transport further increases.
When lactate leaves the cell, it may be taken up by liver, heart, kidneys or other tissues where it may be oxidized or used for gluconeogenesis. However, with severe hypoxia or ischemia, lactate production outpaces clearance by liver and kidneys, so lactate and H⁺ accumulate in blood. Lactate itself is a strong anion; at physiological pH it is fully dissociated and thus lowers blood pH.
Acidosis and Cell Dysfunction:
Acidosis causes cell dysfunction mainly by disturbing protein structure, ion gradients, and energy metabolism, which then impair signaling, contraction, and survival. The tertiary conformation of proteins and enzymes are dependent on protonation of amino acids. Amino acids with basic side chains, (lysine, arginine and histidine) are protonated at physiological pH, while acidic amino acids aspartate and glutamate are protonated in low pH environments. With changes in optimal protein conformation, enzyme activity is often reduced. These findings are of heightened significance in regards to energy homeostasis, as low pH impairs mitochondrial respiratory capacity and thus ATP production. Subsequently, cell ATP-dependent ion pump dysfunction may result in a loss of cell membrane potential and volume regulation, and ultimately cell death. Specifically, reduced extracellular pH inhibits neuronal voltage-gated and ligand-gated ion channels, allowing calcium and sodium to enter the cells. The resulting cell depolarization and neuronal excitation activates cellular injury pathways. In addition to ion channel effects, downstream protein binding to acid-sensing ion channels (ASIC) activates cell death pathways (apoptosis, necroptosis). Ironically, a critical component of the neuronal cell death pathway is a serine/threonine kinase receptor interaction protein 1, which is aptly named RIP1. Intracellular acidosis also interferes with many pH-sensitive processes, including metabolism, motility, immune function and growth, in multiple cell types (e.g., neurons, astrocytes, colon, skeletal muscle, macrophage). Specifically impacted pathways which regulate these functions include MAPK, PI3K-Akt-mTOR as well as TRAIL–death receptor signaling. Thus, prolonged acidosis may shift cells from adaptation toward apoptosis or necrosis. Without question acidosis itself is a putative neural injury factor.
Hypoxia and Cell Dysfunction:
Similar to metabolic acidosis, hypoxia causes cell dysfunction primarily by limiting mitochondrial ATP production, altering gene expression, disturbing ion homeostasis, and activating cell death pathways. Hypoxia primarily injures cells through oxygen lack and energy failure, while metabolic acidosis mainly alters pH-sensitive proteins, ion transport, and signaling, even when oxygen is present. The mechanism of response to hypoxia involves hypoxia inducible factors (HIFs), which are stabilized by low oxygen availability and control the expression of a multitude of genes, including those involved in cell survival, angiogenesis, and glycolysis. If persistent and/or severe, hypoxia may produce secondary metabolic acidosis via anaerobic glycolysis and lactate accumulation; thus, many hypoxic injuries combine energy failure with pH-mediated enzyme and ion-transport disturbances (see above). However, mild acidosis may transiently reconfigure mitochondrial efficiency to maintain mitochondrial function and cell survival. Thus, mild extracellular acidosis actually may be a protective agent, but severe or prolonged acidosis worsens dysfunction and promotes cell death.
Hypercapnia (Elevated CO2) and Cell Dysfunction:
Although fetal/newborn metabolic acidosis is often accompanied by hypercapnia, the biochemical and physiologic consequences of hypercapnia, as well as the interaction with acidosis and hypoxia, are far less studied than that of acidosis or hypoxemia. Whereas metabolic acidosis disturbs enzyme activity, ion gradients and muscle contractility, hypercapnia changes cell function via both CO2 itself and via respiratory acidosis, as CO2 hydration to carbonic acid causes respiratory acidosis, lowering intra- and extracellular pH. As compared to modest physiologic or pathologic increases in pCO2 under conditions of hypoventilation or increased CO2 production in children and adults, fetal/newborn umbilical artery blood may demonstrate far greater pCO2 levels, at times exceeding 100 mmHg. Adverse labor events resulting in absent umbilical placental clearance (e.g., complete cord compression, total abruption, uterine rupture) results in an increase of pCO2 of ~7 mmHg per minute. As metabolic acidosis (i.e., base deficit) increases ~0.5 mmol/L per minute under these conditions, newborns with severe acidosis most often present with a mixed respiratory and metabolic acidosis. The preferential diffusion of bicarbonate ion (resulting from CO2 hydration) as compared to H+ from the blood to the extracellular compartment mandates the accurate calculation of base deficit with correction for this extracellular exchange (Base Deficit extracellular fluid).
Similar to metabolic acidosis, elevated CO2 reduces mitochondrial oxygen consumption, membrane potential, and electron transport chain activity, leading to lower ATP output from oxidative phosphorylation. Elevated CO2 also may downregulate metabolic pathway enzymes, blunting hypoxia-driven upregulation of glycolytic enzymes and glucose transporters. Criteria for acute hypoxia induced encephalopathy, and thus risk for cerebral palsy (CP), are commonly defined as umbilical artery or newborn pH <7.0 and/or base deficit >12 mmol/L. Metabolic acidosis is much more strongly linked to fetal brain injury than isolated respiratory acidosis from hypercapnia alone, though the pH criteria suggests that respiratory acidosis may be contributive. A 10 minute interruption of umbilical blood flow in a previously normal fetus would predictably increase umbilical artery pCO2 to ~120 mmHg, but only increase base deficit to levels of 7 to 9 mmol/L, values below the base deficit threshold for HIE injury. This would result in an umbilical artery pH ~6.91, placing the infant within the high risk category based upon pH.
There is limited data regarding the risk for CP based on predominant respiratory acidosis with sub-injury BD threshold values, as the occurrence is uncommon. Tuuli et al demonstrated that umbilical cord arterial lactate (as a measure of metabolic acidosis) was a more discriminating measure of term infant neonatal morbidity than was pH, though other studies indicate that umbilical cord arterial lactate/base excess and pH predict short-term neonatal outcomes with similar efficacies.
In view of the diverse metabolic effects, the duration of metabolic and/or respiratory acidosis potentially also may impact on the risk of neonatal metabolic injury. Umbilical cord artery hypercarbia is typically corrected within minutes of life with adequate neonatal ventilation. In contrast, newborns do not significantly clear metabolic acidosis until beyond 1 ½ to 2 hours, due to immaturity of hepatic and renal mechanisms at birth. Thus, the cellular effects of metabolic acidosis are likely to impact far longer than that of acute hypercarbia. Importantly, efforts to chemically normalize metabolic acidosis (e.g., bicarbonate administration) have not demonstrated efficacy, but may, in fact, result in adverse effects. For example, intravenous alkali infusions to acidotic dogs induced cerebral vasoconstriction and reduced cerebral blood flow. However, with ischemic stroke-induced brain acidosis, treatment with NaHCO3 significantly reduced infarct volume. Despite the potential cellular effects, it is likely that severe metabolic acidosis creates a markedly greater risk of neurologic injury than does hypercapnia. However, there may well be an interplay between the effects of hypoxia, metabolic acidosis and respiratory acidosis.
Brain Regions Impacted by Fetal Hypoxemia and Acidosis
Classic patterns of fetal hypoxic-ischemic brain damage have differentiated Acute-Profound hypoxia patterns from that of Partial-Prolonged hypoxic damage. The former, a consequence of sudden sentinel events (e.g., abruptio, cord prolapse, uterine rupture) often cause symmetric injury to the basal ganglia and thalami (deep gray nuclei) and sometimes brainstem involvement. In contrast, Partial-Prolonged events often damage parasagittal watershed regions between major cerebral arteries, with relative sparing of deep nuclei. Depending upon the severity and mechanism of injury (fetal hemorrhage vs hypoxia-ischemia) other affected regions may include the hippocampus, brainstem and cerebellum.
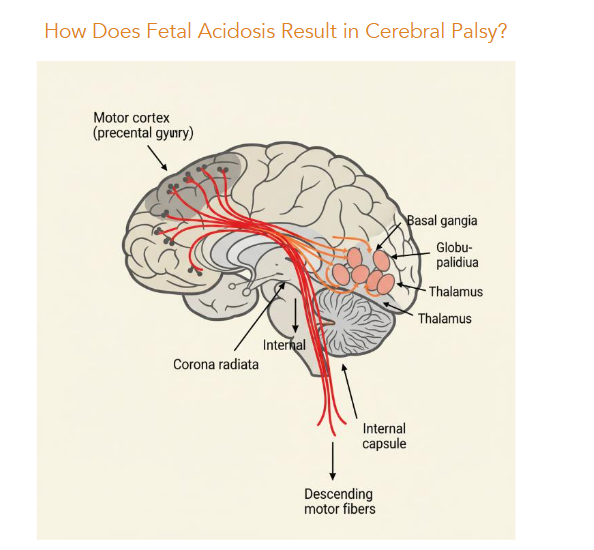
The susceptibility of the basal ganglia to acute hypoxemia is believed to be due to the high metabolic demand, dense excitatory input, and vascular features that limit their reserve during sudden drops in oxygen or perfusion. Having a high resting metabolic rate, the basal ganglia depend on continuous oxidative ATP production; even brief interruptions in oxygen may rapidly exhaust energy stores. The basal ganglia neurons receive massive glutamatergic input from cortex and thalamus; during hypoxia–ischemia, energy failure impairs glutamate reuptake and increases synaptic glutamate, over activating NMDA/AMPA receptors with the excitotoxic cascade leading to Ca++ overload.
Watershed regions lie at the junction of two of the three major cerebral arterial territories (anterior, middle and posterior). As such, being at the farthest, lowest-pressure ends of arterial territories and having the poorest collateral supply, sustained decreases in oxygen delivery (perfusion or oxygenation) may drop below thresholds necessary to prevent injury.
Acidosis Thresholds for Cerebral Palsy
As noted above, to attribute hypoxic-ischemic encephalopathy and cerebral palsy (CP) to an intrapartum event requires values of umbilical artery or newborn pH <7.0 and/or base deficit >12 mmol/L, or both. As pH is an inverse log of the hydrogen ion concentration, the relation with the degree of acidosis is logarithmic. Linear changes in umbilical artery pH are of limited value in predicting the severity of injury, as pH combines the degree of metabolic acidosis with the often transient respiratory acidosis. In contrast, base deficit is a linear measure of the degree of metabolic acidosis, and base deficit extracellular fluid corrects for the effect of CO2 levels. Base deficit measures indicate a linear or exponential correlation of the degree of metabolic acidosis with the risk of neonatal death, though a threshold phenomenon for the risk of cerebral palsy.
Among infants with severe acidosis, the rate of death (up to 2 years of age) increased from 5% at BD 12-15.9 mmol/L, to 10% at 16-19.9 mmol/L, to 26% at BD -20 mmol/L. In contrast, among infants with a BD of 12-15.9 mmol/L, the rate of cerebral palsy was 2.1%, which did not increase significantly (4.0%) among those with BD 16-19.9 mmol/L, while there was a marked increase (33%) in the rate of cerebral palsy at >20 mmol/L.
The apparent threshold levels of 12 and 20 mmol/L may be reflective of the complex mechanisms by which acidosis results in neurologic injury as well as the contribution from fetal/newborn cardiovascular impairment (i.e., increased ischemia) which occurs at the highest levels of acidosis.
Therapeutic Approaches to Newborn Hypoxic Injury:
Certainly, prevention of fetal hypoxic-ischemic injury prenatally or intrapartum in the ultimate objective. However, should severe acidosis result, current pediatric care approaches have had significant success in preventing or reducing the degree of damage. Certainly, quality resuscitation and stabilization may limit the hypoxic event. Therapeutic hypothermia (whole-body or head cooling) of nearterm or term infants with moderate–severe hypoxic-ischemic encephalopathy reduces death and severe disability and lowers rates of cerebral palsy compared with normothermia. Therapeutic hypothermia efficacy is focused on interrupting the delayed injury cascade that unfolds hours to days after the initial asphyxial event. Cooling the brain reduces cerebral metabolic rate, helping preserve ATP and delay or blunt the secondary energy failure that normally peaks 12–48 hours after hypoxia–ischemia.
Hypothermia also dampens glutamate release, NMDA/AMPA receptor activation, and downstream calcium-dependent enzyme activation, thereby limiting excitotoxic damage in vulnerable regions. Cooling also lowers production of reactive oxygen and nitrogen species and stabilizes mitochondrial membranes, attenuates microglial activation and pro-inflammatory cytokine release, and interferes with apoptotic pathways.
Pharmacologic adjuncts to therapeutic hypothermia have attempted to address the multiple pathways of neuronal damage induced by hypoxia, acidosis and hypercapnia, including energy production and utilization, excitatory stimulation, inflammatory cytokines, reactive oxygen species and apoptotic pathways. Studies of agents including erythropoietin, melatonin, allopurinol, magnesium sulfate, xenon, topiramate, and mesenchymal stem cells show potential promise, but demonstrated benefit has not been confirmed.
Conclusion
Fetal/newborn hypoxic-ischemic brain injury is a consequence of direct cellular effects of hypoxia coupled with secondary effects of metabolic acidosis and, potentially, that of hypercapnia. The complexity of mechanisms inducing neuronal damage include altered protein structure, abnormal ion gradients, reduced cellular energy metabolism, increased excitatory neuronal signals, inflammatory cytokines, activation of apoptotic pathways, and reoxygenation injury. Major questions remain unanswered, including what factors predict infant tolerance/susceptibility to severe metabolic acidosis, the relative impact of acidosis vs. hypoxemia and hypercapnia, and whether persistence of acidosis during the newborn period impacts cellular injury. Whereas therapeutic hypothermia likely acts broadly to reduce cellular energy requirements and suppress damage pathways, the identification of these putative damage pathways indicates opportunities for therapeutic pharmacologic targets.
Conflict of Interest Statement: None.
Funding Statement: None.
Acknowledgements: AI assistance (Perplexity) was used to assist in writing. All AI-information and AI-suggested references were checked for validity. One Figure (brain detail) was generated with AI.
References:
- Burton, B.K., Inborn errors of metabolism in infancy: a guide to diagnosis. Pediatrics, 1998. 102(6): p. E69.
- Rabinowitz, J.D. and S. Enerbäck, Lactate: the ugly duckling of energy metabolism. Nat Metab, 2020. 2(7): p. 566-571.
- Li, X., et al., Lactate metabolism in human health and disease. Signal Transduct Target Ther, 2022. 7(1): p. 305.
- Genders, A.J., et al., A physiological drop in pH decreases mitochondrial respiration, and HDAC and Akt signaling, in L6 myocytes. Am J Physiol Cell Physiol, 2019. 316(3): p. C404-c414.
- Chu, X.P., et al., Modulation of acid-sensing ion channels: molecular mechanisms and therapeutic potential. Int J Physiol Pathophysiol Pharmacol, 2011. 3(4): p. 288-309.
- Wang, Y.Z., et al., Tissue acidosis induces neuronal necroptosis via ASIC1a channel independent of its ionic conduction. Elife, 2015. 4.
- Salameh, A.I., V.A. Ruffin, and W.F. Boron, Effects of metabolic acidosis on intracellular pH responses in multiple cell types. Am J Physiol Regul Integr Comp Physiol, 2014. 307(12): p. R1413-27.
- Degitz, C., S. Reime, and O. Thews, Effect of Acidosis-Induced Signalling Pathways on Mitochondrial O(2) Consumption of Tumour Cells. Adv Exp Med Biol, 2022. 1395: p. 231-235.
- Cheng, H., et al., Extracellular acidosis restricts one-carbon metabolism and preserves T cell stemness. Nat Metab, 2023. 5(2): p. 314-330.
- Hagelund, S. and A. Trauzold, Impact of Extracellular pH on Apoptotic and Non-Apoptotic TRAIL-Induced Signaling in Pancreatic Ductal Adenocarcinoma Cells. Front Cell Dev Biol, 2022. 10: p. 768579.
- Solaini, G., et al., Hypoxia and mitochondrial oxidative metabolism. Biochim Biophys Acta, 2010. 1797(6-7): p. 1171-7.
- Lee, P., N.S. Chandel, and M.C. Simon, Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat Rev Mol Cell Biol, 2020. 21(5): p. 268-283.
- Trollmann, R. and M. Gassmann, The role of hypoxia-inducible transcription factors in the hypoxic neonatal brain. Brain Dev, 2009. 31(7): p. 503-9.
- Khacho, M., et al., Acidosis overrides oxygen deprivation to maintain mitochondrial function and cell survival. Nat Commun, 2014. 5: p. 3550.
- Bing, O.H., W.W. Brooks, and J.V. Messer, Heart muscle viability following hypoxia: protective effect of acidosis. Science, 1973. 180(4092): p. 1297-8.
- Ross, M.G., Forensic Analysis of Umbilical and Newborn Blood Gas Values for Infants at Risk of Cerebral Palsy. J Clin Med, 2021. 10(8).
- Olofsson, P., Umbilical cord pH, blood gases, and lactate at birth: normal values, interpretation, and clinical utility. Am J Obstet Gynecol, 2023. 228(5s): p. S1222-s1240.
- Berend, K., Diagnostic Use of Base Excess in Acid-Base Disorders. N Engl J Med, 2018. 378(15): p. 1419-1428.
- Vohwinkel, C.U., et al., Elevated CO(2) levels cause mitochondrial dysfunction and impair cell proliferation. J Biol Chem, 2011. 286(43): p. 37067-76.
- Fergie, N., et al., Hypercapnic acidosis induces mitochondrial dysfunction and impairs the ability of mesenchymal stem cells to promote distal lung epithelial repair. Faseb j, 2019. 33(4): p. 5585-5598.
- Reddan, B. and E.P. Cummins, The regulation of cell metabolism by hypoxia and hypercapnia. J Biol Chem, 2025. 301(3): p. 108252.
- Baalbaki, S.H., et al., Predicting long-term neurodevelopmental outcomes in very preterm neonates by umbilical cord gas parameters. Am J Obstet Gynecol MFM, 2021. 3(1): p. 100248.
- Tuuli, M.G., et al., Umbilical cord arterial lactate compared with pH for predicting neonatal morbidity at term. Obstet Gynecol, 2014. 124(4): p. 756-761.
- Einikyte, R., et al., The comparison of umbilical cord arterial blood lactate and pH values for predicting short-term neonatal outcomes. Taiwan J Obstet Gynecol, 2017. 56(6): p. 745-749.
- Victory, R., et al., Umbilical cord pH and base excess values in relation to adverse outcome events for infants delivering at term. Am J Obstet Gynecol, 2004. 191(6): p. 2021-8.
- Shah, P.S., et al., Recovery of metabolic acidosis in term infants with postasphyxial hypoxic-ischemic encephalopathy. Acta Paediatr, 2003. 92(8): p. 941-7.
- Al-Shehri, H., et al., The practices of intravenous sodium bicarbonate therapy in neonatal intensive care units: A multi-country survey. Medicine (Baltimore), 2023. 102(29): p. e34337.
- Lokesh, L., et al., A randomized controlled trial of sodium bicarbonate in neonatal resuscitation-effect on immediate outcome. Resuscitation, 2004. 60(2): p. 219-23.
- Thuo, E., E.R. Lyden, and E.S. Peeples, Effect of early clinical management on metabolic acidemia in neonates with hypoxic-ischemic encephalopathy. J Perinatol, 2024. 44(8): p. 1172-1177.
- Arvidsson, S., E. Häggendal, and I. Winsö, Influence on cerebral blood flow of infusion of sodium bicarbonate during respiratory acidosis and alkalosis in the dog. Acta Anaesthesiol Scand, 1981. 25(2): p. 146-52.
- Pignataro, G., R.P. Simon, and Z.G. Xiong, Prolonged activation of ASIC1a and the time window for neuroprotection in cerebral ischaemia. Brain, 2007. 130(Pt 1): p. 151-8.
- de Vries, L.S. and F. Groenendaal, Patterns of neonatal hypoxic-ischaemic brain injury. Neuroradiology, 2010. 52(6): p. 555-66.
- Johnston, M.V., et al., Neurobiology of hypoxic-ischemic injury in the developing brain. Pediatr Res, 2001. 49(6): p. 735-41.
- Baburamani, A.A., et al., Vulnerability of the developing brain to hypoxic-ischemic damage: contribution of the cerebral vasculature to injury and repair? Front Physiol, 2012. 3: p. 424.
- Beltz, E.E. and M.E. Mullins, Radiological reasoning: hyperintensity of the basal ganglia and cortex on FLAIR and diffusion-weighted imaging. AJR Am J Roentgenol, 2010. 195(3 Suppl): p. S1-8 (Quiz S9-11).
- Wang, B., et al., Neuroprotection in the Striatum of Hypoxic-Ischemic Piglets by Simultaneous Inhibition of Dopamine D1 and Adenosine A2A Receptors. Neonatology, 2022. 119(3): p. 354-360.
- Chacko, A., et al., Cortical ischaemic patterns in term partial-prolonged hypoxic-ischaemic injury-the inter-arterial watershed demonstrated through atrophy, ulegyria and signal change on delayed MRI scans in children with cerebral palsy. Insights Imaging, 2020. 11(1): p. 53.
- Kelly, R., et al., Dose-dependent relationship between acidosis at birth and likelihood of death or cerebral palsy. Arch Dis Child Fetal Neonatal Ed, 2018. 103(6): p. F567-f572.
- Ross, M.G., Threshold of metabolic acidosis associated with newborn cerebral palsy: medical legal implications. Am J Obstet Gynecol, 2019. 220(4): p. 348-353.
- Shankaran, S., Therapeutic hypothermia for neonatal encephalopathy. Curr Treat Options Neurol, 2012. 14(6): p. 608-19.
- Gonzalez, F.F., Neuroprotection Strategies for Term Encephalopathy. Semin Pediatr Neurol, 2019. 32: p. 100773.
- Gunn, A.J., et al., Therapeutic hypothermia translates from ancient history in to practice. Pediatr Res, 2017. 81(1-2): p. 202-209.
- Toro-Urrego, N., et al., Neuroprotective Role of Hypothermia in Hypoxic-ischemic Brain Injury: Combined Therapies using Estrogen. Curr Neuropharmacol, 2019. 17(9): p. 874-890.
- González-Ibarra, F.P., J. Varon, and E.G. López-Meza, Therapeutic hypothermia: critical review of the molecular mechanisms of action. Front Neurol, 2011. 2: p. 4.
- Molloy, E.J., et al., Neuroprotective therapies in the NICU in term infants: present and future. Pediatr Res, 2023. 93(7): p. 1819-1827.