

Inhibition of ACC1 Reduces Malignancy in Hepatobiliary Cancers
Inhibition of ACC1 Diminishes the Malignant Phenotype of Hepatobiliary Cancers In Vitro
Noel Jacquet1, Jianhua Yu2, Xinggui Shen3, Yunfeng Zhao1
- Department of Pharmacology, Toxicology & Neuroscience, LSU Health Sciences Center, Shreveport, LA 71130, USA.
- Department of Hepato-Biliary-Pancreatic Surgery, Shaoxing People’s Hospital, Shaoxing, Zhejiang, China.
- Department of Pathology and Translational Pathobiology, LSU Health Sciences Center, Shreveport, LA 71130, USA.
OPEN ACCESS
PUBLISHED: 30 November 2024
CITATION: Jacquet, N., Yu, J., et al., 2024. Inhibition of ACC1 Diminishes the Malignant Phenotype of Hepatobiliary Cancers In Vitro. Medical Research Archives, [online] 12(11).
https://doi.org/10.18103/mra.v12i11.5759
COPYRIGHT © 2024 European Society of Medicine. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
DOI https://doi.org/10.18103/mra.v12i11.5759
ISSN 2375-1924
Abstract
Hepatobiliary cancers are a collection of malignancies arising from liver and biliary tract cells. These cancers have high anatomical proximity, overlapping symptoms, and share common risk factors. Hepatobiliary cancers are generally rare and often lack effective treatment options due to late diagnoses and limited treatment efficacy. These cancers tend to be highly aggressive, which limits treatment options for patients, especially since diagnosis often occurs at later stages of disease progression. Chronic inflammation is the most significant risk factor for developing these malignancies. Inflammation alters cellular metabolism, which gives them unique metabolism characteristics that enhance their survival and increase malignancy. Acetyl-Coa-carboxylase (ACC) is a metabolic enzyme responsible for the carboxylation of acetyl-CoA (AC) into malonyl-CoA (MC). MC is used in de novo fatty acid synthesis, an upregulated process in human cancers. Acetyl-Coa-carboxylase 1 (ACC1), in particular, is the first rate-limiting step for de novo lipogenesis. Here we delve into the effect of ACC1 inhibition on the malignant phenotypes of hepatobiliary cancers. Our results showed that knockdown of ACC1 slowed proliferation and migration, reduced spheroid formation, and altered cell cycle progression and protein expression in hepatobiliary cancers. In conclusion, our study suggests that ACC1 may contribute to hepatobiliary cancers’ malignancy and may be utilized as a therapeutic target for treating such diseases.
Keywords
hepatobiliary cancers, Acetyl-Coa-carboxylase, ACC1, cancer metabolism, malignant phenotype
Introduction
Hepatobiliary cancers are a collection of malignancies arising from liver and biliary tract cells. Biliary tract cancers include cholangiocarcinoma and gallbladder cancers. Gallbladder cancer (GBC) is the most common type of biliary tract cancer. It is often characterized as being highly aggressive, which results in poor prognosis for diagnosed individuals. In our previous studies, we demonstrated how the metabolic protein UCP2 regulates malignant cellular traits, such as proliferation, chemoresistance, and cellular plasticity in GBC. Incidence rates of gallbladder cancer are on the rise, particularly in women, South American, and South Asian populations. About 30% of the surface of the gallbladder is attached to the bottom surface of the liver and empties the cystic duct into the common hepatic duct, forming the common bile duct. This interconnected pathway allows for high levels of metastasis between these organs.
The biliary tree connects canaliculi and bile ducts from the liver and gallbladder. Malignancies in the bile ducts or other parts of the biliary system are known as cholangiocarcinoma (CCA). The incidence rates of cholangiocarcinoma are similarly rising worldwide in trend with other hepatobiliary cancers. The intrahepatic and extrahepatic ducts branch from within the liver throughout the rest of the biliary tree to form the common hepatic duct. Dysfunction along any portion of the biliary tree can impact metabolism throughout other parts of the system. The intricate connection of this system leads to high vulnerability among patients suffering from hepatobiliary cancers. The development of these cancers can quickly impact a patient’s overall health as their metabolic functions rapidly begin to decline.
Hepatocellular carcinoma (HCC) is a type of liver cancer with a rising global incidence rate, especially in African American and Native American populations. HCC is the third leading cause of cancer-related deaths globally and has one of the highest death rates when compared to its incidence rates. Previous studies from our lab have explored the interaction between HCC and CCA, focusing on how the presence of CCA cells can induce malignant traits, such as epithelial-mesenchymal transition (EMT), in HCC cells. HCC arises from hepatocytes, the primary cell type in the liver. These cells are responsible for several metabolic functions, including drug metabolism, protein synthesis, and carbohydrate processing. These cells also produce bile which flows through canaliculi and is used throughout the biliary tree in metabolic processes.
Metabolic diseases such as diabetes and obesity can serve as risk factors for hepatobiliary cancers, especially HCC. Several cancer hallmarks- including alterations to cellular metabolism- are responsible for the malignant phenotype of these cancers, which make them incredibly complex and difficult to treat. Tumorigenesis depends greatly on reprogramming an organism’s cellular metabolism. The liver has an essential role in managing the body’s metabolism, which includes energy production, de novo lipid synthesis, and the breakdown of xenobiotics. Cancer cells have the ability to thrive in nutrient-deficient environments. They will often hijack the body’s existing resources to feed their development. Mutations tend to alter gene expression, cellular differentiation, and cellular environment. This can lead to cancer-induced inflammation and the alteration of metabolic processes.
Fatty acid synthesis is critical to cellular metabolism and several other physiological and pathological conditions. Malonyl-CoA (MA) is used by fatty acid synthase to produce long-chain saturated fatty acids. Acetyl-CoA carboxylases (ACCs) are enzymes that catalyze the carboxylation of acetyl-CoA into malonyl-CoA. ACC1 is the primary member of the ACC family in mammals. These enzymes are vital to the biosynthesis of fatty acids; they are the rate-limiting step of the reaction. Several metabolic processes rely on pools of malonyl-CoA, including fatty acid oxidation in the mitochondria. As a result, the activity of ACC1 indirectly regulates these processes.
Acetyl-Coa-carboxylase 1 plays a pivotal role in many diseases, such as cancer, diabetes, and obesity. Studies have shown there to be a link between ACC1 expression and disease promotion in obesity, diabetes, and NAFLD. ACC1 has been shown to be highly expressed in tissues with high metabolic activities like lipogenesis; this includes the liver. Evidence suggests that ACC1 is upregulated in several human cancers; this promotes lipogenesis, which is required for the rapid growth and proliferation of cancer cells. ACC1 dysregulation has been linked to regulating tumor cell proliferation and migration. This has so far been observed in breast and prostate cancers and liposarcomas. This article aims to study the role of ACC1 in Hepatobiliary cancers using cell lines from three major types of cancers.
Hepatobiliary cancers often have a high rate of metastasis between each other which is often enhanced by the tumor microenvironment. These cancers have high mutation rates which leads to their increased proliferation and metastasis. The symptomology in all three cancers is indistinct from many other health conditions which frequently results in delayed diagnosis for patients. Late diagnosis limits the availability for treatment options and often corresponds to worse disease progression. Currently, there is a lack of screening tools for hepatobiliary cancers. Patients typically only undergo screening after being diagnosed with a chronic liver disease like hepatitis or cirrhosis or in the rare case of family history. Our work is looking to highlight the potential role of ACC1 to serve as a biomarker and therapeutic target for hepatobiliary cancers.
Hepatobiliary cancers are highly aggressive malignancies with rising global incidence rates. The interconnected nature of these organs facilitates a high level of metastasis which contributes to poor overall survival for patients with GBC, CCA, and HCC. The tumorigenesis of these malignancies is closely linked to changes in cellular metabolism. ACC1 is a metabolic enzyme vital for the production of malonyl-CoA and fatty acid synthesis. Lipid synthesis particularly is highly expressed in metabolic tissues like the liver and its dysregulation has been linked to the tumorigenesis of several human cancers. The liver and biliary tree are highly sensitive to alterations in these metabolic pathways which supports evidence to investigate ACC1 as a therapeutic intervention for these cancers. This study focuses on the role of ACC1 in hepatobiliary cancers by exploring its effects in cell lines from GBC, CCA, and HCC to better understand its contribution to cancer progression.
Materials and Methods
1. CELL CULTURE AND REAGENTS
G415 (GBC), HuCCT-1 (CCA), and SNU-449 (HCC) cells were grown in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% Fetal Bovine Serum (FBS), 1% L-glutamine, 0.4% penicillin, and 0.2% Plasmocin. Each cell line was incubated at 37 ͦC in 5% CO2. ACC1 shRNA CRISPR lentivirus was purchased from Applied Biological Materials Inc (ABM).
2. DATA ANALYSIS OF HUMAN HEPATOBILIARY CANCERS
Human hepatobiliary cancer data was obtained from the Gene Expression Profiling Interactive Analysis (GEPIA). We generated overall survival analysis plots for ACC1 high and low expression groups among HCC and CCA patients, using a median cutoff and a 95% confidence interval. ACC1 expression box plots for HCC and CCA tumor tissues were also created with GEPIA software, applying a p-value threshold of 0.01 and a Log2FC cutoff of 1.
3. DETECTION OF ACC1 mRNA LEVELS IN LIVER CANCER PATIENTS SAMPLE
Tissue samples were obtained from 13 patients at the Shaoxing People’s Hospital from January 2019 to December 2019. Informed consent was obtained from patients, and the tissue acquisition protocol was approved by the Ethics Committee of Shaoxing People’s Hospital. These patients were diagnosed with liver cancer according to pathological examination. Among them, 11 patients were diagnosed with hepatocellular carcinoma, and two patients were diagnosed with intrahepatic cholangiocarcinoma. Both tumor tissues and tumor-adjacent normal tissues were obtained for our study. Tissue samples were ground with cooling by liquid nitrogen. Total RNAs were isolated from tissues using TRIzol (Invitrogen) and reverse transcribed into cDNAs using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s instructions. After, SYBR Green PCR Master Mix was added, a quantitative real-time PCR was performed using the ABI 7500 Real-time PCR system (Applied Biosystems). The designed PCR primers were as followings: ACC1 forward primer, 5′-TTCACTCCACCTTGTCAGCGGA-3′; ACC1 reverse primer, 5′- GTCAGAGAAGCAGCCCATCACT-3′; β-actin forward primer, 5′-AGAAGGAGATCACTGCCCTGGCACC-3′; β-actin reverse primer, 5′-CCTGCTTGCTGATCCACATCTGCTG-3′. β-actin was used as an endogenous control. Relative expression levels were determined using the 2−ΔΔCT methods.
4. GENERATION OF ACC1 STABLE KNOCKDOWN CELLS
G415, HuCCT-1, and SNU-449 cells were cultured and transfected using an ACC1 shRNA CRISPR lentivirus and control vectors purchased from Applied Biological Materials Inc. Seventy-two hours post-infection cells underwent puromycin selection for two weeks. Resistant colonies were selected and amplified for future use. Western blot analysis was performed to evaluate ACC1 expression in scramble siRNA controls and to assess the reduction of ACC1 expression in knockdown clones. Clone generation is produced similarly to what we have described in previous publications.
5. PROLIFERATION INCUCYTE ASSAY
SNU-449, G415, and HuCCT-1 cells (2,500 in 200 µl growth medium) were seeded into 96-well plates (Corning, cat log #353072). Plates were placed in the IncuCyte Zoom cell incubator (Sartoris). Proliferation rates were observed and calculated in real time via the Incucyte software. Images were taken every four hours at 10 x magnification for 120 hours.
6. WOUND HEALING ASSAY
HuCCT-1 and G415 cells (50,000 cells in 200 µl growth medium) were seeded into 96-well plates (Essen, cat log # 4379) and incubated overnight. The next day, wounds were created using a wound maker tool. Plates were then placed in the IncuCyte Zoom incubator, and images were taken every four hours at 10x magnification for 96 hours.
7. 3D SPHEROID FORMATION ASSAY
For 3D spheroid assays, SNU-449, G415, and HuCCT-1 cells were seeded into 96-well ultra-low attachment plates (Costar, cat log # 7007) at 5,000 cells per well. Normal culture media was supplemented with 1% Matrigel. Plates were centrifuged at 300 x g for 5 minutes and placed into the IncuCyte Zoom incubator. IncuCyte software was used to take images every four hours at 10 x magnification for three days.
8. CELL CYCLE ANALYSIS
For our cell cycle analysis HuCCT-1, G415, and SNU-449 cells were harvested via trypsinization. Cells were fixed in ice-cold 70% ethanol. The ethanol was aspirated out to prepare for staining, and the cells were washed twice with 1mL PBS. FxCycle PI/RNAse solution was added to each sample, and the samples were covered with foil and left to develop for 30 minutes at room temperature. Samples were analyzed using flow cytometry.
9. WESTERN BLOT ANALYSIS
Cell lines were harvested by trypsinization or cell scrapping from culture flasks. Cell pellets were then resuspended in RIPA buffer and sonicated on ice for three strokes, ten seconds each. Samples were then incubated on ice for 30 min. After incubation, samples were centrifuged at 4 ͦ C at 18,000 g for 30 min. The supernatant was collected and designated as the whole cell lysate, which was used for Western blot analysis. The whole cell lysate was quantified, and an equal amount of proteins was separated on 12% SDS-page gels. Proteins were transferred onto PVDF membranes. The membranes were blocked using 2% non-fat milk prepared in PBS-tween. An ECL solution (Prometheus ProSignal, Genesee Scientific) was used for imaging under a ChemiDoc imaging system (Bro-Rad).
10. PROTEOMIC ANALYSIS
Proteomic analysis was performed by collecting SNU-449, G415, and HuCCT-1 cells that were lysed using EasyPep Lysis Buffer (Thermofisher Cat log#A45735), and the protein concentrations were detected by a BCA assay. Protein (10 µg) from each sample was loaded into SDS-PAGE and stained with Imperial Protein Stain. The Gel lanes were cut into 2mm X 2mm cubes, and then trypsin was digested, followed by disulfide bond reduction with dithiothreitol and cysteine alkylation with iodoacetamide, according to the manufacturer’s protocol of the Thermo Scientific™ Gel Tryptic Digestion kit (Thermofisher Cat log#89871). The digested peptides were vacuum-dried and resolved in the LC/MS grade water with 0.1% (v/v) formic acid, followed by untargeted discovery proteomics analysis. This LC-MS/MS analysis was carried out using an Ultimate 3000 RSLCnano system connected to an Orbitrap Exploris 480 mass spectrometer. The digested peptides (5.0 µl) were loaded onto a trap column (PepMap C18, 2 cm × 100 μm, 100 Å) at a flow rate of 20 μl/min using 0.1% formic acid, and separated on an analytical column (EasySpray 50 cm × 75 μm, C18 1.9 μm, 100 Å) with a flow rate of 300 nl/min with a linear gradient of 5 to 45% solvent B (100% ACN, 0.1% formic acid) over a 120 min gradient. Both precursor and fragment ions were acquired in the Orbitrap mass analyzer. Precursor ions were acquired in the m/z range of 375-1500 with a resolution of 120,000 (at m/z 200). Precursor fragmentation was carried out using the higher-energy collisional dissociation method using normalized collision energy (NCE) of 32. The fragment ions were acquired at a resolution of 150,000 (at m/z 200). The scans were arranged using a top-speed method with a 3-second cycle time between MS and MS/MS. Ion transfer capillary voltage was maintained at 2.1 kV. The raw mass spectrometry data were analyzed using Proteome Discover (version 2.5, Thermo Fisher Scientific) software package with SequestHT using species-specific fasta database and the Percolator peptide validator. Cysteine alkylation was set as a fixed modification. Phosphorylation on serine/threonine/tyrosine residues and deamidation of asparagine residues were selected as variable modifications.
11. STATISTICAL ANALYSIS
Statistical analysis was performed using Prism 7. Experiments were repeated at least three times. Data represent the mean ± standard deviation (SD), and the differences between groups were analyzed using One-way ANOVA followed by the Tukey–Kramer adjustment or Student’s t-test. p< 0.05 was defined as significantly different.
Results
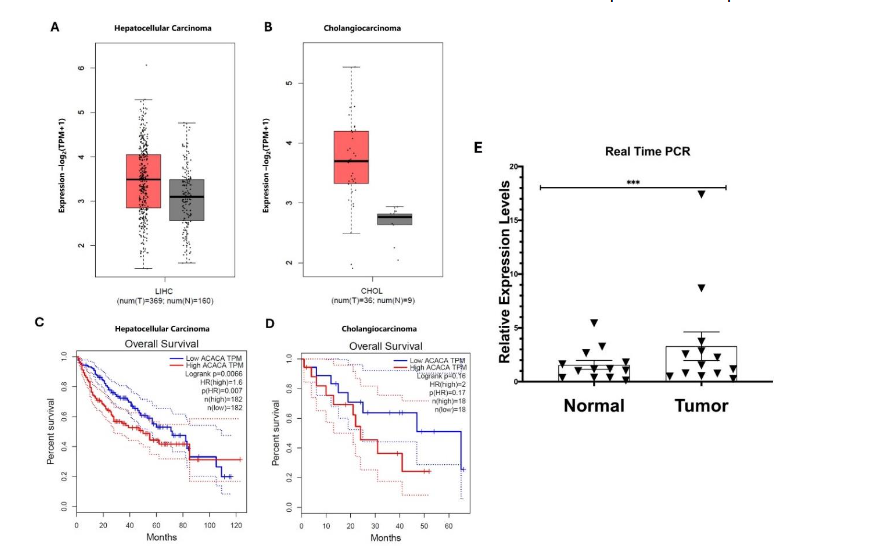
ACC1 Was Highly Expressed in Hepatobiliary Cancers and Associated with Poor Patient Survival
Using the Gene Expression Profiling Interactive Analysis 2 (GEPIA) tool, we analyzed ACC1 expression and overall survival rates of patients with hepatocellular carcinoma (HCC) and cholangiocarcinoma (CCA) (Figure 1). The GEPIA database revealed that ACC1 was expressed at higher levels in tumor tissues compared to the adjacent normal tissue in both HCC (Figure 1A) and CCA (Figure 1B). Elevated ACC1 expression in patients with hepatocellular carcinoma (Figure 1C) and cholangiocarcinoma (Figure 1D) was associated with poorer overall survival.
We performed a preliminary study using 13 pairs of liver cancer tumor/normal tissues and detected the mRNA expression of ACC1. As shown in Figure 1E, the expression levels of ACC1 were highly expressed in tumor tissues compared to the adjacent normal tissues.
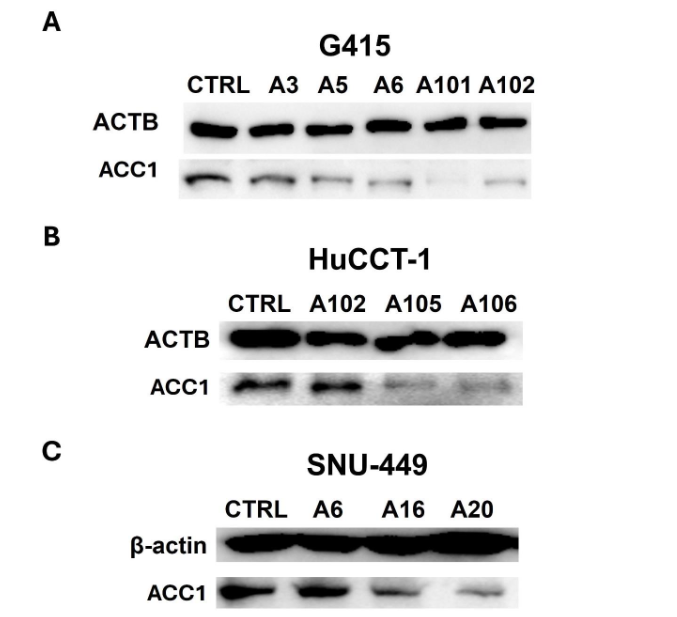
ACC1 Inhibition Suppressed the Proliferation of Hepatobiliary Cancer Cells.
Since ACC1 is highly expressed in hepatobiliary cancers, we generated ACC1 stable knockdown clones of G415 (gallbladder cancer), HuCCT-1 (cholangiocarcinoma), and SNU-449 (hepatocellular carcinoma) cells to study whether inhibition of ACC1 could suppress the growth of these cancer cells. After selection, viable clones were analyzed via Western blot analysis, which showed a reduction in ACC1 expression in each cancer type. The clone selection results are shown in Figure 2.
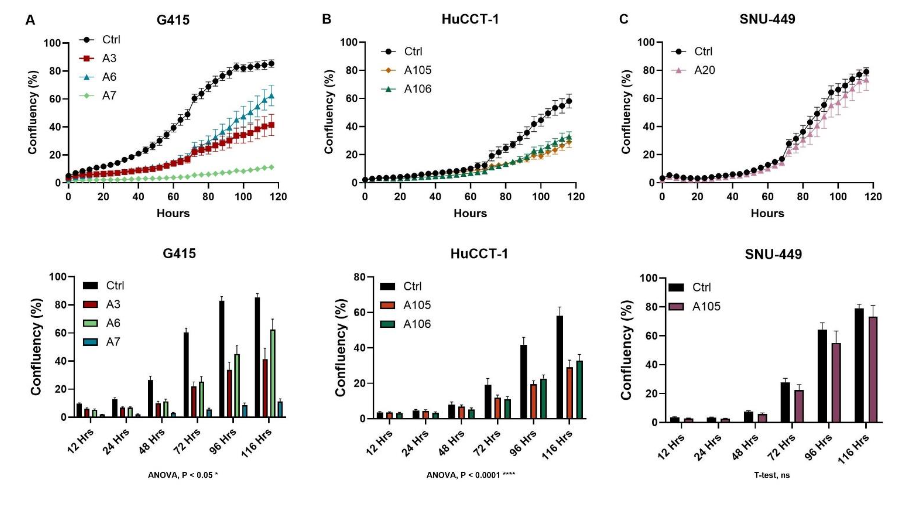
To study how ACC1 affects the proliferation of hepatobiliary cancer cells, we performed an IncuCyte proliferation assay on control and ACC1 knockdown clones (Figure 3). Cell growth was observed for 120 hours. The proliferation rates in the ACC1 knockdown cells were much slower in the G415 (Figure 3A) and HuCCT-1 cells (Figure 3B) but not significantly reduced in SNU-449 cells (Figure 3C).
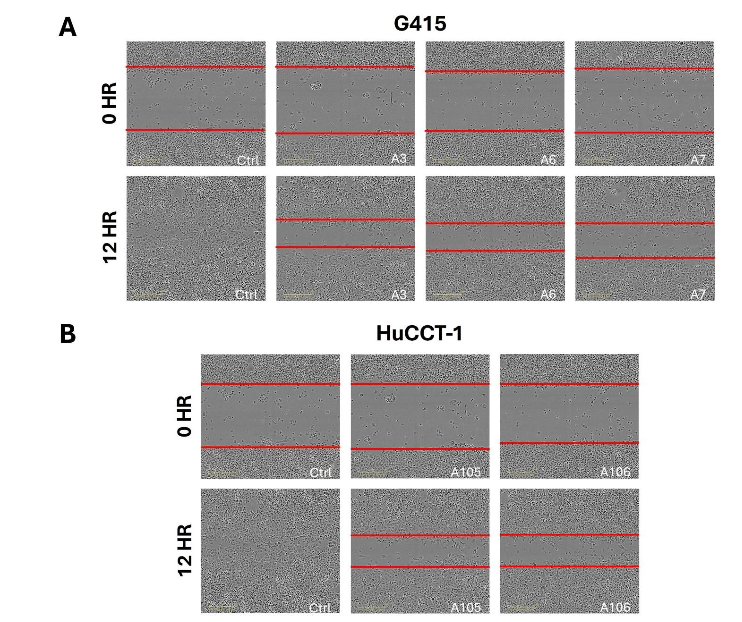
ACC1 Inhibition Suppressed the Migration of Hepatobiliary Cancer Cells.
The scratch wound assay measures migration, another important contributing factor to tumor malignancy. We performed this assay on our G415 and HuCCT-1 cancer control and ACC1 knockdown cells. The rate of migration was slower in the ACC1 knockdown cells compared to the control cells (Figure 4). This effect was seen in the G415 (Figure 4A) and HuCCT-1 clones (Figure 4B).
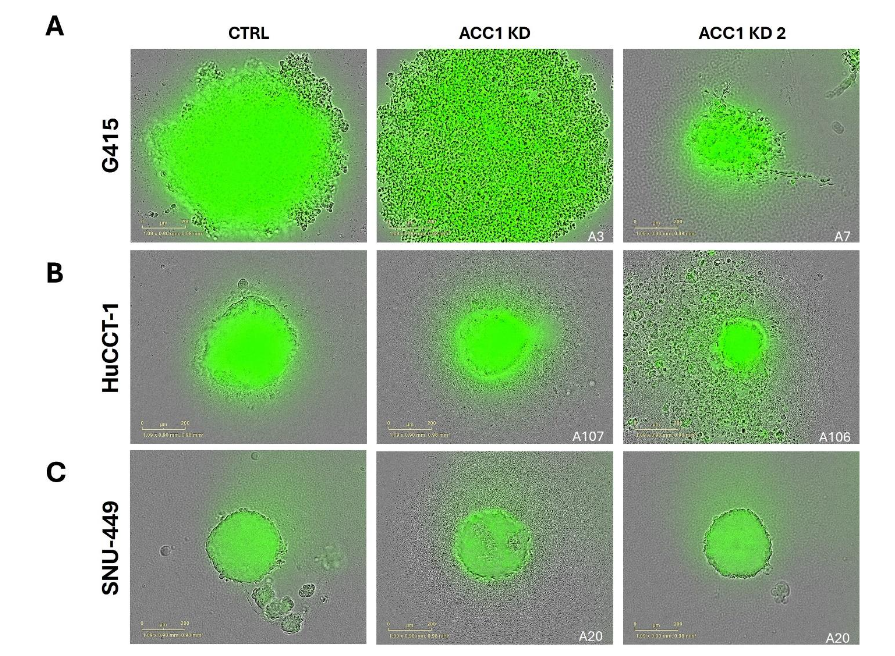
ACC1 Inhibition Suppressed the 3D Growth of Hepatobiliary Cancer Cells.
The formation of 3D spheroids in cell culture mimics tumor growth and survival in vitro. We performed spheroid assays on G415 (Figure 5A), HuCCT-1 (Figure 5B), and SNU-449 (Figure 5C) cells. We observed that the knockdown of ACC1 in these cells reduces the overall cellular density and diameter of spheroids formed in all clones tested except the A3 clone in G415 cells; however, this clone formed looser spheroids.
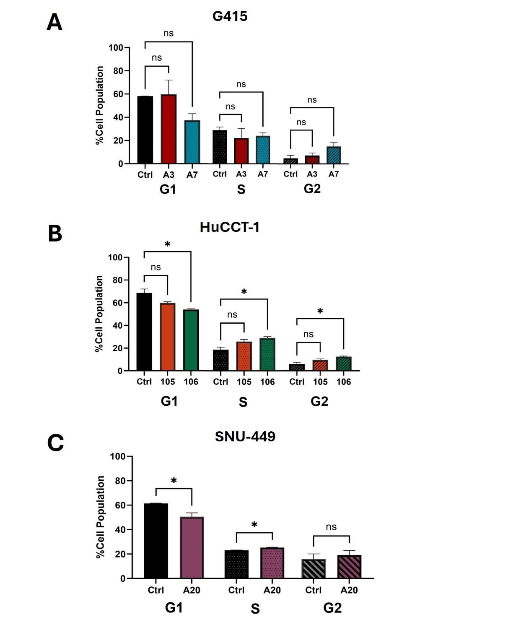
ACC1 Inhibition Altered the Cell Cycle Distribution of Hepatobiliary Cancer Cells
We utilized flow cytometry to analyze the cell cycle population of each cancer cell type. Although there was a trend of changes in cell cycle populations for G415 (Figure 6A), none of the clones were seen to be statistically different from the control cells. The A106-clone of the HuCCT-1 (Figure 6B) cell line showed a significant reduction in the percentage of cells in the G1 phase with a simultaneous increase in the S and G2 phases. The A105-clone trended similarly but was not statistically significant. In the SNU-449 A20-clone ACC1 knockdown clone, there was a significant reduction in the population of cells in the G1 phase, with a simultaneous increase in the S phase (Figure 6C). While not significant, there was also a trend of an increase in population of cells in the G2 phase.
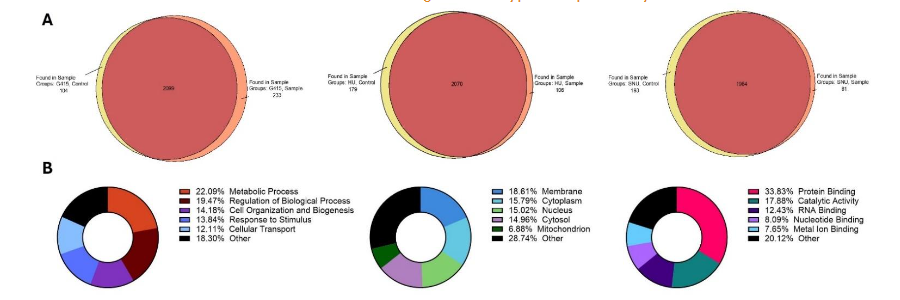
Cellular Processes Altered Due to ACC1 Inhibition in Hepatobiliary Cancer Cells
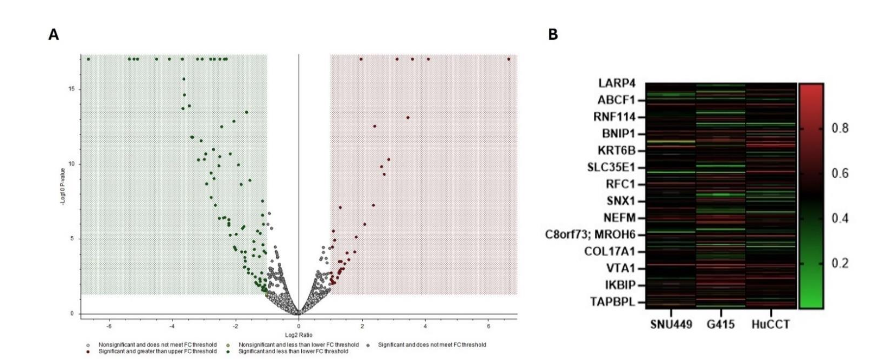
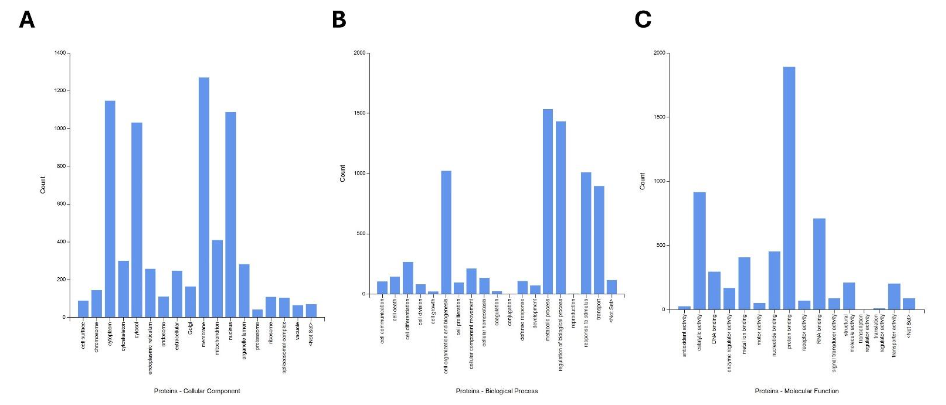
In proteomic analysis conducted on all three cell lines (Figures 7, 8, 9), we observed that the knockdown of ACC1 altered many proteins in each cell line. There were 233 proteins seen to be differentially expressed in the G415 samples, 104 differentially expressed proteins in the HuCCT-1 samples, and 85 differentially expressed proteins in the SNU-449 samples (Figure 7A). Across all three cell lines, there were 377 altered proteins (Figure 8A), 110 of which significantly increased, while 267 proteins were significantly decreased. A heat map of all three cell lines shows the differences in expression for several proteins altered by ACC1 knockdown (Figure 8B). Around one-fifth of the proteins identified were seen to be involved in metabolic processes and the regulation of biological processes (Figure 7B). The most considerable portion of these proteins were seen to be localized in the cellular membrane, cytoplasm, nucleus, or cytosol. Molecular functions affected by the loss of ACC1 include protein binding, catalytic activity, RNA binding, nucleotide binding, and metal ion binding (Figure 7B). The top biological processes these proteins were involved in include metabolic processes, cell organization, response to stimuli, and cellular transport (Figure 9A).
Discussion
Cellular metabolism plays a vital role in tumorigenesis and the malignant phenotype of certain cancers. Hepatobiliary cancers are highly impacted by cellular metabolism, which may contribute to their highly aggressive nature and difficulty in treating. The mechanisms that may make these cancers highly aggressive have yet to be fully elucidated, and the gap in knowledge has led to a lack of treatment options for patients. ACC1 has been shown to be an important regulator of cellular metabolism in several human cancers. In our study, we found that the inhibition of ACC1 in human GBC, CCA, and HCC reduced the malignant phenotype of these cancers. ACC1 expression was seen to be negatively correlated with overall patient survival, and found that ACC1 was highly expressed in HCC tumor samples.
The knockdown of ACC1 was seen to slow the proliferation rates of GBC, CCA, and HCC cancer cells. The reduction in growth was seen across all cell lines, which suggests that ACC1 expression may influence proliferation in cancer cells through mechanisms that need further study. We also observed that knocking down ACC1 in GBC and CCA slowed the migration rate in these cancer cells. Migration is essential for EMT and invasion in cancer. Our data suggests that ACC1 expression may influence this process. Spheroid formation in vitro models tumor formation. Our study observed that ACC1 inhibition decreases spheroids’ cellular density and diameter for GBC, CCA, and HCC cell lines. Alterations in cell cycle progression are common in highly malignant cancers. We see that the reduction of ACC1 in CCA cells significantly influences the population of cells in each cell cycle phase, with similar but non-significant trends in GBC and HCC cells.
Data from our proteomic analysis shows that metabolic processes were the number one pathway influenced by ACC1 expression. Proteins in these pathways are vital for regulating cellular energetics- which influences cancer progression. Cellular processes, including RNA binding and catalytic activity, are also heavily involved in the tumorigenesis of many types of cancers. The alterations in the expression of these proteins by ACC1 inhibition may mean that ACC1 expression is essential to regulating several metabolic processes in the cells that affect tumorigenesis.
The proteins involved in several biological process analyses reveal that the most significantly affected processes include cell organization and biogenesis, metabolic processes, and cell proliferation. This reinforces our hypothesis that ACC1 is pivotal in controlling cell growth and metabolism, which are essential cancer hallmarks. The high count in the metabolic process category further supports the notion that ACC1 affects essential proteins regulating energy production and utilization, thus contributing to the malignant progression of these cancers.
In the molecular function category, catalytic activity and protein binding represent the most abundant functions altered by ACC1 expression, followed by nucleotide binding and DNA binding. These findings suggest that many of the proteins regulated by ACC1 have enzymatic roles that may directly affect metabolic pathways and cell cycle progression. This further emphasizes the enzyme’s involvement in cancer energetics and cellular proliferation. The significant number of proteins involved in nucleotide binding and DNA binding suggests an additional role in gene expression regulation and DNA replication, processes that are frequently dysregulated in cancer cells.
Data from the heatmap identified differential expression in several notable genes. Those marked in red, like LARP4, ABCF1, and RNF114, show elevated expression across all three cell lines, suggesting their potential involvement in pathways commonly upregulated in hepatobiliary cancers. Conversely, genes like IKBIP and TAPBPL are marked by green, indicating lower expression levels, which could point to their potential downregulation by loss of ACC1 expression. There are some notable differences between cell lines like SLC35E1 and NEFM that display variable expression between the cell lines, showing high expression in one line (G415) but reduced in others (SNU449), suggesting a potential context-dependent role in tumorigenesis.
Overall, these proteomic data support the hypothesis that ACC1 influences a wide array of proteins, particularly those involved in metabolic regulation, cellular growth, and gene expression, which are critical in the context of hepatobiliary cancer progression. This comprehensive analysis underscores the potential of targeting ACC1 as a therapeutic strategy to disrupt multiple pathways central to tumorigenesis in these aggressive cancers.
In our study, we found that higher expression of ACC1 in HCC cells could be associated with higher metabolism rates and the malignant phenotype of these diseases. Our results show that ACC1 may be vital in several processes related to significant cancer hallmarks that make hepatobiliary cancer highly aggressive, including sustained proliferative signaling, activation of invasion and metastasis, deregulating cellular energetics, and sustained proliferative signaling. Our data suggests that ACC1 expression may influence proliferative signaling, EMT, metastasis, invasion, and cellular metabolism. Further studies are necessary to fully elucidate the mechanisms by which ACC1 contributes to the tumorigenesis of Hepatobiliary cancers. With more research, ACC1 may prove to be a therapeutic target for these cancers.
Conclusion
This study underscores the significant impact of ACC1 knockdown on the malignant phenotype of hepatobiliary cancers. By reducing ACC1 expression in GBC, CCA, and HCC cells, we observed a clear reduction in key malignant behaviors, such as increased proliferation, migration, and spheroid formation—characteristics associated with the aggressive nature of these cancers. Our findings suggest that targeting ACC1 expression could disrupt critical pathways involved in tumorigenesis, offering a potential therapeutic approach. Future studies are needed to further elucidate the mechanisms by which ACC1 influences cancer progression, specifically its role in pathways related to proliferation, epithelial-to-mesenchymal transition (EMT), invasion, and metastasis.
Conflicts of Interest Statement
The authors declare no conflict of interest.
Funding Statement
The study was supported by the startup package to Yunfeng Zhao from LSU Health Sciences Center-Shreveport. This research was supported by Zhejiang Provincial Natural Science Foundation of China under Grant No. LY22H160008.
Acknowledgments
The authors would like to thank Brain Latimer and David Custis at the institutional Research Core Facility for their assistance in using the IncuCyte incubator and for conducting the cell cycle analysis, respectively.
References
- Groen P. Biliary Tract Cancers | New England Journal of Medicine. The New England Journal of Medicine. 1999;Vol. 341 No. 18. doi:10.1056/NEJM199910283411807
- Krasinskas A. Cholangiocarcinoma – ClinicalKey. Surgical Pathology Clinics. 2018;11(2):403-429.
- Gallbladder cancer | Nature Reviews Disease Primers. Accessed August 28, 2024. https://www.nature.com/articles/s41572-022-00398-y
- Pant K, Gradilone SA. Hepatobiliary Cancers: Progress in Diagnosis, Pathogenesis, and Treatment. Technology in Cancer Research & Treatment. Published online May 12, 2022. doi:10.1177/15330338221097203
- Y W, J L, Y X, et al. Prognostic nomogram for intrahepatic cholangiocarcinoma after partial hepatectomy. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2013;31(9). doi:10.1200/JCO.2012.41.5984
- Satake T, Morizane C, Rikitake R, Higashi T, Okusaka T, Kawai A. The epidemiology of rare types of hepatobiliary and pancreatic cancer from national cancer registry. J Gastroenterol. 2022;57(11):890-901. doi:10.1007/s00535-022-01920-5
- Yu J, Shi L, Lin W, Lu B, Zhao Y. UCP2 promotes proliferation and chemoresistance through regulating the NF-κB/β-catenin axis and mitochondrial ROS in gallbladder cancer. Biochem Pharmacol. 2020;172:113745. doi:10.1016/j.bcp.2019.113745
- Yu J, Shi L, Shen X, Zhao Y. UCP2 regulates cholangiocarcinoma cell plasticity via mitochondria-to-AMPK signals. Biochem Pharmacol. 2019;166:174-184. doi:10.1016/j.bcp.2019.05.017
- World Health Statistics. Accessed August 28, 2024. https://www.who.int/data/gho/data/themes/world-health-statistics
- Banales JM, Marin JJG, Lamarca A, et al. Cholangiocarcinoma 2020: the next horizon in mechanisms and management. Nat Rev Gastroenterol Hepatol. 2020;17(9):557-588. doi:10.1038/s41575-020-0310-z
- Song X, Hu Y, Li Y, Shao R, Liu F, Liu Y. Overview of current targeted therapy in gallbladder cancer. Sig Transduct Target Ther. 2020;5(1):1-19. doi:10.1038/s41392-020-00324-2
- Park JH, Pyun WY, Park HW. Cancer Metabolism: Phenotype, Signaling and Therapeutic Targets. Cells. 2020;9(10):2308. doi:10.3390/cells9102308
- Cholangiocarcinoma (bile duct cancer) – Symptoms and causes. Mayo Clinic. Accessed October 2, 2024. https://www.mayoclinic.org/diseases-conditions/cholangiocarcinoma/symptoms-causes/syc-20352408
- Ilyas SI, Gores GJ. Pathogenesis, Diagnosis, and Management of Cholangiocarcinoma. Gastroenterology. 2013;145(6):1215-1229. doi:10.1053/j.gastro.2013.10.013
- Cancer statistics for the year 2020: An overview – PubMed. Accessed October 9, 2024. https://pubmed.ncbi.nlm.nih.gov/33818764/
- Global Cancer Observatory. Accessed August 28, 2024. https://gco.iarc.who.int/en
- Sung H, Ferlay J, Siegel RL, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71(3):209-249. doi:10.3322/caac.21660
- Gravitz L. Liver cancer. Nature. 2014;516(7529):S1-S1. doi:10.1038/516S1a
- Cen W, Li J, Tong C, et al. Intrahepatic Cholangiocarcinoma Cells Promote Epithelial-mesenchymal Transition of Hepatocellular Carcinoma Cells by Secreting LAMC2. J Cancer. 2021;12(12):3448-3457. doi:10.7150/jca.55627
- Almazroo OA, Miah MK, Venkataramanan R. Drug Metabolism in the Liver. Clinics in Liver Disease. 2017;21(1):1-20. doi:10.1016/j.cld.2016.08.001
- Rumgay H, Ferlay J, de Martel C, et al. Global, regional and national burden of primary liver cancer by subtype. European Journal of Cancer. 2022;161:108-118. doi:10.1016/j.ejca.2021.11.023
- Trefts E, Gannon M, Wasserman DH. The liver. Curr Biol. 2017;27(21):R1147-R1151. doi:10.1016/j.cub.2017.09.019
- Cancer-related inflammation. Accessed August 28, 2024. https://air.unimi.it/handle/2434/145688
- Cellular fatty acid metabolism and cancer – PubMed. Accessed August 28, 2024. https://pubmed.ncbi.nlm.nih.gov/23791484/
- Kamp DW, Shacter E, Weitzman SA. Chronic inflammation and cancer: the role of the mitochondria. Oncology (Williston Park). 2011;25(5):400-410, 413.
- Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell. 2011;144(5):646-674. doi:10.1016/j.cell.2011.02.013
- Stine ZE, Schug ZT, Salvino JM, Dang CV. Targeting cancer metabolism in the era of precision oncology. Nat Rev Drug Discov. 2022;21(2):141-162. doi:10.1038/s41573-021-00339-6
- The Heterogeneity of Liver Cancer Metabolism. In: Le A, ed. The Heterogeneity of Cancer Metabolism. Springer International Publishing; 2021:127-136. doi:10.1007/978-3-030-65768-0_9
- Wang Y, Yu W, Li S, Guo D, He J, Wang Y. Acetyl-CoA Carboxylases and Diseases. Front Oncol. 2022;12:836058. doi:10.3389/fonc.2022.836058
- Luo DX, Tong DJ, Rajput S, et al. Targeting Acetyl-CoA Carboxylases: Small Molecular Inhibitors and their Therapeutic Potential. Recent Patents on Anti-Cancer Drug Discovery. 7(2):168-184. doi:10.2174/157489212799972918
- Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models – PubMed. Accessed October 9, 2024. https://pubmed.ncbi.nlm.nih.gov/27643638/
- Schug ZT, Vande Voorde J, Gottlieb E. The metabolic fate of acetate in cancer. Nat Rev Cancer. 2016;16(11):708-717. doi:10.1038/nrc.2016.87
- Chajès V, Cambot M, Moreau K, Lenoir GM, Joulin V. Acetyl-CoA carboxylase alpha is essential to breast cancer cell survival. Cancer Res. 2006;66(10):5287-5294. doi:10.1158/0008-5472.CAN-05-1489
- Razumilava N, Gores GJ. Cholangiocarcinoma. The Lancet. 2014;383(9935):2168-2179. doi:10.1016/S0140-6736(13)61903-0
- Banales JM, Cardinale V, Carpino G, et al. Cholangiocarcinoma: current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat Rev Gastroenterol Hepatol. 2016;13(5):261-280. doi:10.1038/nrgastro.2016.51
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021 Feb 4. doi: 10.3322/caac.21660. Epub ahead of print. PMID: 33538338.
- Liver cancer – Symptoms and causes. Mayo Clinic. Accessed October 2, 2024. https://www.mayoclinic.org/diseases-conditions/liver-cancer/symptoms-causes/syc-20353659
- Liver Cancer: Symptoms, Signs, Causes & Treatment. Cleveland Clinic. Accessed October 2, 2024. https://my.clevelandclinic.org/health/diseases/9418-liver-cancer
- Kamp DW, Shacter E, Weitzman SA. Chronic inflammation and cancer: the role of the mitochondria. Oncology (Williston Park). 2011;25(5):400-410, 413.
- DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2(5):e1600200. doi:10.1126/sciadv.1600200
- Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metabolism. 2016;23(1):27-47. doi:10.1016/j.cmet.2015.12.006
- GEPIA (Gene Expression Profiling Interactive Analysis). Accessed August 28, 2024. http://gepia.cancer-pku.cn/index.html