

Low-Dose Tamoxifen and Raloxifene for Muscular Dystrophy
Low-dose tamoxifen and raloxifene enhance therapeutic effect of ribitol in mouse model of FKRP mutation-related muscular dystrophy
Anthony Blaeser1, Bo Wu1, Morgan Drains1, Molly Holbrook1, Qi Long Lu1
- McColl-Lockwood Laboratory for Muscular Dystrophy Research, Cannon Research Center, Carolinas Medical Center, Atrium Health, Charlotte, NC 28203, USA.
OPEN ACCESS
PUBLISHED: 30 September 2025
CITATION: Blaeser, A., Wu, B., et al., 2025. Low-dose tamoxifen and raloxifene enhance therapeutic effect of ribitol in mouse model of FKRP mutation-related muscular dystrophy. Medical Research Archives, [online] 13(9). https://doi.org/10.18103/mra.v13i9.6921
COPYRIGHT: © 2025 European Society of Medicine. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
DOI https://doi.org/10.18103/mra.v13i9.6921
ISSN 2375-1924
ABSTRACT
Tamoxifen and raloxifene are selective estrogen receptor modulators (SERM) used to treat or reduce the risk of breast cancer as well as for osteoporosis. These drugs act to block estrogen in various tissues while having a pro-estrogen activity in others. Furthermore, they have been shown to have anti-fibrotic and anti-inflammatory properties making them a potential therapeutic option for muscular dystrophy. Previous studies by our lab and others have examined tamoxifen and raloxifene treatments in muscular dystrophy mouse models with success. These include numerous studies in the mdx mouse resulting in improved muscle function and pathology. Studies in our lab demonstrated long-term, dose dependent improvements in muscle function and pathology with daily treatment of up to 50 mg/kg tamoxifen and up to 100 mg/kg raloxifene. Additionally, ribitol has been shown to be effective in mitigating the dystrophic pathology in the FKRP-P448Lneo- mutant mouse, thus providing a potential supplement option for improving the effectiveness of current drugs. Here we examined the effectiveness of low-dose tamoxifen and raloxifene with and without ribitol supplement. FKRP-P448Lneo- mutant mice were treated with 1 mg/kg tamoxifen or 10 mg/kg raloxifene with and without 2 g/kg ribitol daily for 12 months. Muscle function was examined using grip force, treadmill, whole body plethysmography and echocardiogram at 6 and 12 months. Muscle histology and pathology was examined at 12 months. Muscle function improvements, specifically in treadmill distance and grip force, were seen after 6 months of treatment with combined treatment showing further improvement over each drug alone. Improvements were also noted in respiratory and cardiac parameters. Although there was a reduction in improvements after 12 months of treatment the treated animals retained higher function over untreated controls. Furthermore, an increase in alpha-dystroglycan glycosylation was noted in the combination treated groups as well as improvements in fibrosis and central nucleation in all treated groups but more prominent with the combined treatment. Taken together these data suggest the combinatorial treatment is effective over drug alone. These results demonstrate the potential benefit of low dose tamoxifen and raloxifene for the treatment of FKRP-related muscular dystrophies and other muscular dystrophies.
Keywords
tamoxifen, raloxifene, ribitol, FKRP mutation, muscular dystrophy
Introduction
O-manno glycosylation of alpha-dystroglycan (α-DG) is crucial for binding of the membrane to extracellular matrix proteins, maintaining muscle membrane integrity and protecting from contractile induced damage. Dystroglycanopathies are a group of diseases caused by the reduced or lack of α-DG glycosylation (hypoglycosylation) which leads to a loss of muscle cell membrane integrity and repeated cycles of degeneration/regeneration. This process is accompanied by the accumulation of fibrotic and fat tissues in the muscle, leading to loss of function in both cardiac and skeletal muscles. Mutations in the fukutin-related protein (FKRP) gene are one of the major causes of dystroglycanopathies with clinical manifestation ranging from mild LGMD2I to severe congenital muscular dystrophy. Currently, there is no approved treatment for FKRP-related diseases. However, several experimental therapies have been identified by pre-clinical studies in animal models and two of them are under clinical trials. One promising treatment is ribitol administration which has been demonstrated with safety and efficacy in both animal models and in phase I/II clinical trials. Ribitol is a natural metabolite widely present in organisms including mammals although its metabolic pathway has not been clearly understood. Recent studies demonstrate that ribitol can be converted to ribitol-5 phosphate (Rbo5P) by the kinase FGGY and subsequently converted to CDP-ribitol by CDP-Rbo pyrophosphorylase A/isoprenoid synthase domain-containing (CRPAA/ISPD). FKRP has been identified as a Rbo5P transferase, utilizing CDP-ribitol for the addition of Rbo5P to the core glycan chain of α-DG, providing the base necessary for the addition of a repeating disaccharide of xylose (Xyl) and glucuronic acid (GlcA) (matriglycan) required for the binding of the extracellular matrix proteins. Increase in the substrate CDP-ribitol enhances the efficiency of mutant FKRP function and, consequently, the levels of matriglycan. It has now been demonstrated that ribitol treatment indeed can increase levels of Rbo5P and CDP-ribitol in muscle tissues and is associated with improved muscle pathology and function in disease relevant mouse models with FKRP mutation. Studies by our lab using the FKRP-P448Lneo- (P448L) mutant mouse model have demonstrated recovery of matriglycan synthesis following ribitol treatment. Ribitol treatment dose dependently increases matriglycan and improves muscle pathology and function. A clinical trial is also underway to test ribitol in LGMD2I patients (NCT05775848) with positive Phase 2 results reported including improved matriglycan expression in muscles, improved serum CK and sustained 10 meter walk test (MLBio/BridgeBio results). No severe side effects have been identified. The safety and effectiveness of ribitol as a therapeutic for FKRP-related muscular dystrophy provide a potential means of supplementing other therapeutics such as gene or drug therapies to increase their effectiveness and possibly lower the required dose.
One of the pathogenic processes within dystrophic muscle is the persistent inflammation initiated by muscle degeneration. An inflammatory response is essential for normal skeletal muscle repair, but the chronic inflammatory environment impairs muscle regeneration, exacerbates the loss of muscle mass and accelerates accumulation of fibrotic and fat tissues within diseased muscles. The process of fibrosis associated with inflammation involves many factors including transforming growth factor (TGF-β), which is activated chronically in dystrophic muscles, and induces the deposition of fibrotic proteins such as collagen replacing degenerated muscle fibers. Therefore, suppressing the inflammatory response and fibrosis has been the primary attempt as a potential therapy for muscular dystrophies in general, and remains one of the most practiced treatments despite well-recorded side effects and limited efficacy. This line of drugs includes glucocorticoids, such as deflazacort, prednisone, and Vamorolone (VBP15).
Selective estrogen receptor modulators (SERMs) act on estrogen receptors (ERs) either as ER agonists or antagonists and induce a wide range of effects in a tissue and cell type specific manner. This is because estrogen receptors, as either a or b form, are widely present in nearly all tissues, but at different levels and in variable proportions. The use of SERMs therefore results in a broad spectrum of effects on functions of nearly all systems by differentially altering ERα and ERβ actions. SERM drugs have long been explored for treating diseases and its therapeutic effect have been well established for the prevention of breast cancer, bone loss and against inflammation and fibrosis. Other benefits include improvement to muscle repair and enhancements of muscle function likely through stabilizing biological membranes and inhibition of apoptosis. These effects are considered desirable for treating muscular dystrophies with several reports of animal model trials of tamoxifen.
Our laboratory earlier examined the use of tamoxifen and raloxifene for their therapeutic potential in the P448L mutant mouse. The mice were treated with tamoxifen and raloxifene at doses ranging from either 2, 10 and 50 mg/kg/day or 50 and 100 mg/kg/day respectively for 12 months. Treatments of each drug resulted in improved grip strength as well as running time and distance via treadmill exercise. Furthermore, enhanced cardiac and respiratory function were noted as well as a reduction in bone loss. These positive effects are apparently dose-dependent. No significant adverse effects were noted in the animals treated with 50 mg/kg raloxifene. However, tamoxifen treatment causes severe adverse effects on male reproductive organs and hernia with varying degree in severity, dose-dependently. Specifically, tamoxifen at the dose of 50 mg/kg greatly reduced the masses of vas deferens and testicles with seminal vesicles hardly visible. Reduction in size of these organs becomes less severe, but remained detectable even at the dose of 2 mg/kg. Therefore, while our results and those reported in animal models of other muscular dystrophies clearly suggest therapeutic potentials of tamoxifen for muscular dystrophy, the risks with high dose of the drug could well hinder its clinical application, especially for muscular dystrophies which requires life-long treatment.
Complementary mechanisms of action offered by SERMs and ribitol prompted us to examine the therapeutic values of a combined treatment of the SERMs with ribitol. In this study, we tested the long-term efficacy of tamoxifen and raloxifene at doses lower than the human equivalent in clinics in P448L mutant mice.
Methods
ANIMALS.
The use of animals in this study was approved by the Atrium Health/Wake Forest Institutional Animal Care and Use Committee, Atrium Health Wake Forest School of Medicine (Charlotte, NC). All mice were housed in the vivarium at the Carolinas Medical Center following animal care and guidelines of the institute.
ANIMALS, DRUGS, AND IN VIVO DELIVERY METHODS
The P448L mutant mouse was used with C57Bl/6 mice as controls. The P448L mutant mouse contains a c.1343C>T point mutation resulting in an amino acid change from proline to leucine at the 448 position. In each treatment group, 10 P448L or C57BL/6 mice (five female and five male mice), aged 8 weeks, were used. The mice were randomly assigned to control and different treatment groups, with littermates split between control and treatment groups.
Treated P448L mice were given either 1 mg/kg tamoxifen (Letco Medical, Decatur, AL) or 10 mg/kg raloxifene (Teva Pharmaceuticals, North Wales, PA) daily, 6 days per week, for 12 months via oral gavage with or without 2 g/kg ribitol (Biosynth, Gardner, MA). Control P448L mice were gavaged with the same amount of saline. Mice were sacrificed at scheduled time points, and muscles and organs snap frozen in cold isopentane (−80°C) and stored at −80°C. All attempts were made to analyze experiments blind to the treatment groups.
HISTOLOGY
As described previously, tissue sections (6 μm thick) were stained with monoclonal antibody IIH6C4 (Millipore, Temecula, CA) and Alexa Fluor 594–labelled goat–anti-mouse IgM (Invitrogen, Eugene, OR) for detection of functionally glycosylated α-DG. Sections were stained with hematoxylin and eosin or Masson’s trichrome for histologic assessment of fiber size and centranucleation using the MetaMorph Basic Offline software version 7.7.0.0 (Molecular Devices LLC, Sunnyvale, CA). The percentages of fibrosis in diaphragm, heart, and biceps muscles were measured from Masson’s trichrome staining (ImageJ software version 1.42; NIH, Bethesda, MD; http://imagej.nih.gov/ij). The level of serum components was determined by Charles Riverside Laboratories International (Wilmington, MA).
PROTEIN EXTRACTION AND WESTERN BLOT ANALYSIS
Total proteins were extracted from each muscle shaving using TX-100 buffer [1% Triton X-100, 50 mmol/L Tris (pH 8.0), 150 mmol/L NaCl, and 0.1% SDS] supplemented with protease inhibitor cocktail (Roche, Mannheim, Germany). Samples were homogenized in TX-100 buffer, and the supernatants were collected by centrifugation at 16,000 × g for 10 minutes. Protein concentration was determined by Bradford assay (Bio-Rad DC protein assay). The lysates were then loaded onto 4% to 20% Tris-glycine gel (Invitrogen, Carlsbad, CA). The proteins were transferred to polyvinylidene difluoride membranes with constant ampere at 200 mA for 2 hours in cold room (4°C). Polyvinylidene difluoride membranes were incubated with protein-free T20 blocking buffer (Pierce, Rockford, IL). The antibodies against α-DG (IIH6C4) and α-actin (Sigma, St. Louis, MO) were incubated in 20 mmol/L Tris (pH 7.4), 150 mmol/L NaCl, and 0.1% Tween 20 at 1:2000 dilutions. α-DG and α-actin antibodies were detected by horseradish peroxidase–goat anti-mouse IgM (Invitrogen, Carlsbad, CA) and goat anti-rabbit IgG–horseradish peroxidase conjugate (Bio-Rad, Hercules, CA), respectively. Blots were developed with electrochemiluminescence (PerkinElmer, Waltham, MA), and the images were exposed and processed by an LAS-4000 imaging system (Fujifilm, Valhalla, NY).
GRIP STRENGTH AND TREADMILL TEST
Grip strength was measured using a horizontal forelimb mesh and hindlimb angled mesh grip strength meter (Columbus Instruments, Columbus, OH). Five successful forelimb and hind limb strength measurements were recorded within 2 minutes with the highest and lowest measurements removed. Data was averaged and normalized to body weight (BW).
Treadmill test was performed on LE8700 treadmill (Panlab/Harvard Apparatus, Barcelona, Spain). Mice were acclimated with a 0-degree plane for 5 minutes at a speed of 7 cm/second. After the acclimation the speed was increased by 1cm/second every 30 seconds until a maximum speed of 25 cm/second was reached. A 0.2-mA shock grid was used throughout the procedure for motivation. Mice were considered exhausted if they remained on the shock grid for 10 consecutive seconds without getting off or 50% on/off within 1-minute period. Mice were also monitored for any signs of distress or injury and removed promptly if any signs appeared.
PLETHYSMOGRAPHY AND ECHOCARDIOGRAPHY
Whole body plethysmography was performed using the Emka Technologies Whole Body Plethysmograph with IOX Respiratory function analysis software (Emka Technologies, Falls Church, VA). Mice were acclimated to individual chambers for 5 minutes prior to recording respiratory data. Immediately following the acclimation period the data was collected for 15 minutes with continuous exchange of air.
Echocardiography was performed as previously described by Blaeser et al using the BioScan SonixTablet High Frequency Ultrasound (Analogic Ultrasound, Peabody, MA).
STATISTICAL ANALYSIS
All of the results were expressed as means ± SEM. The data were analyzed using two-tailed t-test to compare tamoxifen- or raloxifene-treated groups with untreated controls. P ≤ 0.05 is considered to be significant.
Results
1. Recovery of glycosylated α-DG after 12 months of treatment with tamoxifen and raloxifene.
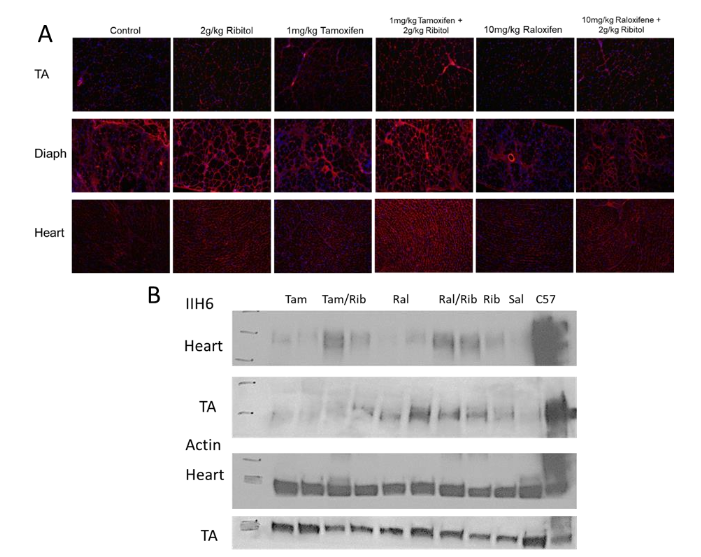
Hypoglycosylation of α-DG is the key characteristic of FKRP-related dystroglycanopathies and a primary therapeutic target. The glycosylation is detected using the IIH6 antibody with affected muscle of P448L mutant mice showing little to no IIH6 staining, except for those regeneration-related revertant fibers. We examined glycosylation in the TA, heart and diaphragm of control and treated mice to determine if treatment with low doses of tamoxifen (1 mg/kg) or raloxifene (10 mg/kg) could enhance ribitol-induced matriglycan synthesis. We’ve previously demonstrated restoration of α-DG glycosylation in FKRP mutant mice within a range of doses of ribitol. P448L mice treated with 2 g/kg ribitol showed an increase in glycosylation, especially in the diaphragm and heart tissues, consistent with our previous results. Treatments with 1 mg/kg tamoxifen and 10 mg/kg raloxifene alone did not show significant restoration of α-DG glycosylation. Combined treatments of tamoxifen or raloxifene with ribitol showed a substantial increase in IIH6 staining in TA, diaphragm and heart (Figure 1A). This is confirmed by Western blot (Figure 1B). However, signal intensity in matriglycan is not clearly different between the combined treatment compared to ribitol alone.
2. Histological analysis of P448L mutant mice following tamoxifen and raloxifene treatment.
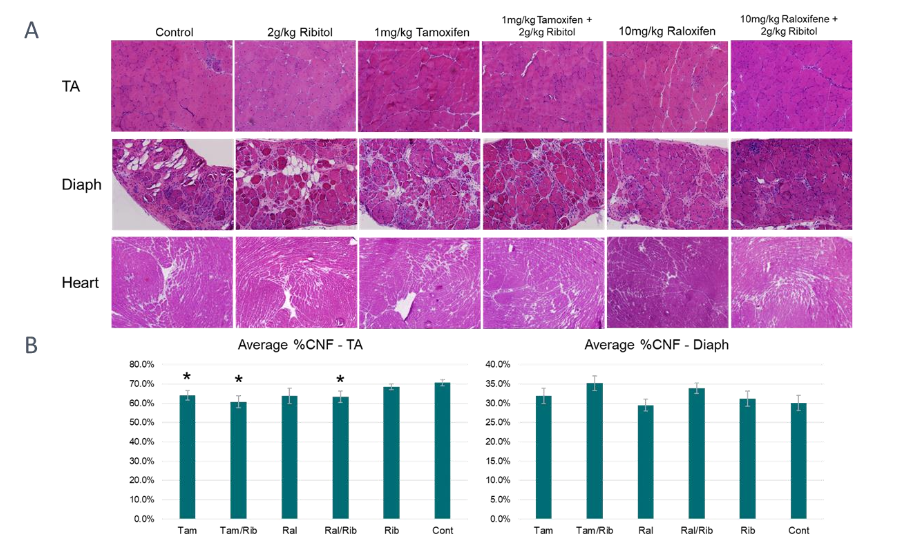
A hallmark of pathology in the P448L mutant mouse is an increase in centrally nucleated fibers (CNF) due to repeated degeneration and regeneration. These CNFs are of various sizes and tend to be in clusters signifying regeneration. Control P448L mice had an average CNF of ~71% in the TA. Our earlier study showed that treatments with 2 mg/kg Tamoxifen and 50 mg/kg Raloxifene demonstrate a small, but statistically significant decrease in the number of CNF in the TA. Treatment with 1mg/kg tamoxifen or 10 mg/kg raloxifene, with and without ribitol, for 1 year showed a similar reduction in CNF in the TA. A small reduction in the number of CNF was seen in the ribitol treated mice (68%) with a further reduction in raloxifene (68%) with statistically significant reductions in tamoxifen (64%) and raloxifene/ribitol combination (63%) therapies. The largest, statistically significant, reduction was seen in the tamoxifen/ribitol group with a reduction in CNF to less than 61% (Figure 2). On the other hand, only a small reduction in centrally nucleated fibers was seen in the diaphragm of raloxifene treated animals with 30% CNF compared to control mice with 31%. Ribitol treatment resulted in similar CNF levels with approximately 31%. However, an increase in CNF was seen in the raloxifene/ribitol treatment with 34% CNF and a further increase in tamoxifen and tamoxifen/ribitol both resulting in CNF of 35%. It should be noted that none of the changes in CNF percentage in the diaphragm are statistically significant.
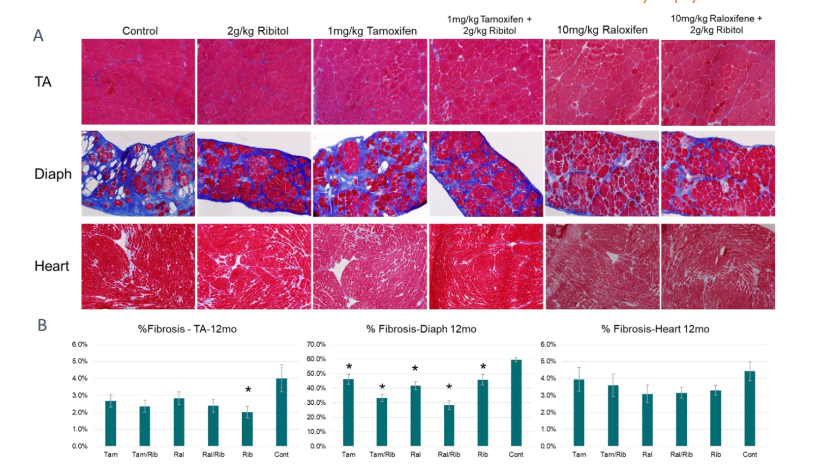
The amount of fibrotic area was reduced with the treatment of SERMs and ribitol. (Figure 3). Fibrotic area consisted of approximately 4% of the tissue in the control TA muscles and was reduced by all the treatments especially the groups treated with tamoxifen/ribitol, raloxifene/ribitol and ribitol alone, down to 2.4, 2.4 and 2.0% of the tissue area respectively, However, statistical significance is only observed between the ribitol treated and the control group. The heart of control animals has limited fibrotic tissue, approximately 4.5% area. A reduction is detected in the treated groups with 4.0% in the tamoxifen alone and 3.6% in the tamoxifen combination groups. A further reduction is noted in the other three treatment groups with just above 3% fibrosis in both raloxifene treatment groups and ribitol alone, though not statistically significant. The progressive fibrosis is most evident in the diaphragm of the aged control P448L mouse accounting for about 60% total area of the tissue. Treatment with tamoxifen and raloxifene for 12 months with and without ribitol show a significant reduction in fibrosis. down to less than 50%. Interestingly, the combination treatments showed the most improvement with a reduction in fibrosis down to 33% and 28% in the tamoxifen/ribitol and raloxifene/ribitol treatment groups, respectively.
3. Evaluation of muscle function by treadmill and grip force
Muscle function was assessed twice, at 6- and 12-months post-treatment. Treadmill exercise showed that all the treatments improve performance when compared to the control P448L mice. Improved performance in both the mice treated with ribitol alone and tamoxifen alone was statistically significant in running distance at 12-month over the control. Furthermore, significant improvement was detected in both running distance and time at both time points in the mice treated with tamoxifen in combination with ribitol. Interestingly, treatment with raloxifene alone also significantly improved both running distance and time at both time points. Moreover, combined treatment of raloxifene and ribitol presented the most significant functional improvement, more than that achieved in the raloxifene alone at the 12 month time point, and with running distance and time almost doubled that achieved by the control mice (Figure 4). It should also be noted that the overall weight of the animals was only slightly less in the tamoxifen groups compared to control animals. However, animals of both raloxifene groups are significantly lighter at both 6-month and 12-month time points. The association of significant improvement in muscle function with lower body weight is consistent with our earlier study of higher dose SERM treatment in the same mouse model. These results together suggest that improvement in muscle functions may be linked to maintenance of proper body weight, or body weight itself can be an indicator of general health of the mice.
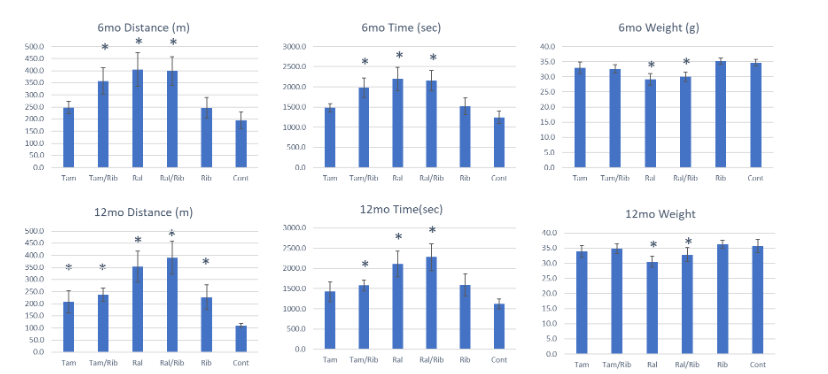
Measurement of muscle functions at 2 time points provides data for direct comparison of the same individual and groups of animals in treadmill performance over time. While there is a considerable variation between animals, a significant (~43%) reduction in performance at 12-month point was recorded when compared to that at 6-month point in the control mice. A much smaller reduction in performance was seen between the two time points in the tamoxifen treated groups, ~33% and 16% with and without ribitol, respectively. A minimal reduction was observed in the group with ribitol alone (~7%) especially in the raloxifene-treated groups, ~13% and only ~2% without and with ribitol respectively. Also interesting is the change in body weight (BW) between control and treated groups. Control mice showed an increase of approximately 6.5% from 6 to 12 months post-treatment, while ribitol treated mice showed a smaller increase of ~3.4%. Tamoxifen treated mice also increased weight by ~5% and ~3% with and without ribitol, respectively. Interestingly, the mice treated with raloxifene alone and in combination gained just under 5% weight between 6-month and 12-month time points. However, BW increase was much slower during the first 6 months of raloxifene treatment. As a results, the raloxifene groups maintained an overall lower weight compared to all the other groups. It should also be noted that the raloxifene groups did not appear to be underweight as the average weight at 6 months post-treatment was ~30g with raloxifene alone and ~33g with combined treatment with ribitol after 12 months of treatment.
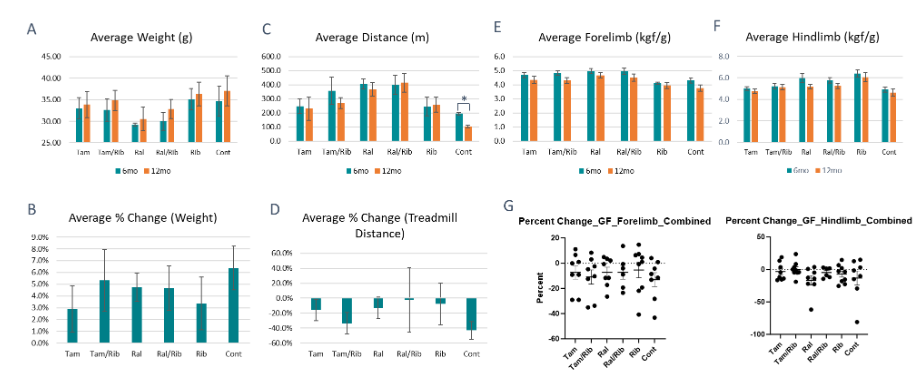
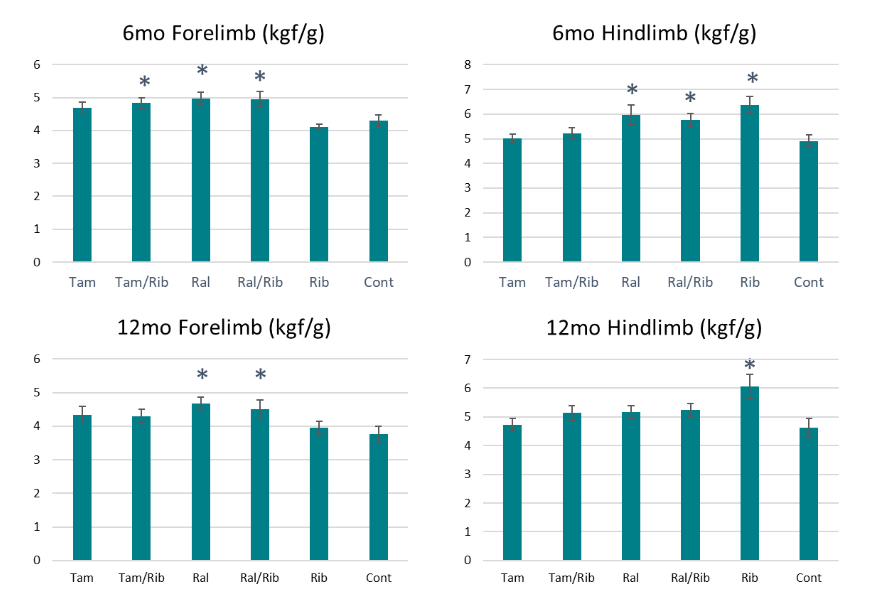
The improvement of the treatments on muscle functions was also demonstrated by grip strength measurement. Forelimb and/or hindlimb grip strength were improved at 6-month time point in all treatment groups, and significance was observed for forelimb strength for the groups with combination treatment and raloxifene alone. Hindlimb strength was significantly improved only in the groups treated with raloxifene and ribitol alone. These results are similar to the treadmill tests with both raloxifene treatment groups showing significant improvements over control. Significant improvement is maintained in hindlimb strength with ribitol treatment alone and in forelimb strength with raloxifene treated groups at 12-month compared to the control mice. However, improvements are not statistically significant in all other treatment groups.
We also compared percent change in grip force between 6 and 12 month treatment points. Control mice showed a drop in strength of ~6.5% in the hindlimb and ~13% in the forelimb. A similar reduction in forelimb grip strength was seen with Tamoxifen with and without ribitol and with raloxifene alone. A drop of 12% in hindlimb force was also observed with raloxifene alone. However, reduction in hindlimb was much less in the tamoxifen groups (1% and 5% with and without ribitol respectively) than that of the control. Ribitol alone showed a reduction of only 4% and 5% in the forelimb and hindlimb, respectively. Interestingly, there is only a loss of ~4% forelimb strength and ~5% hindlimb strength in the raloxifene combination treatment allowing for the treated mice to maintain a grip strength above controls. Overall, the functional data demonstrate efficacy with all the treatments and raloxifene groups present the most consistent improvements by both functional tests.
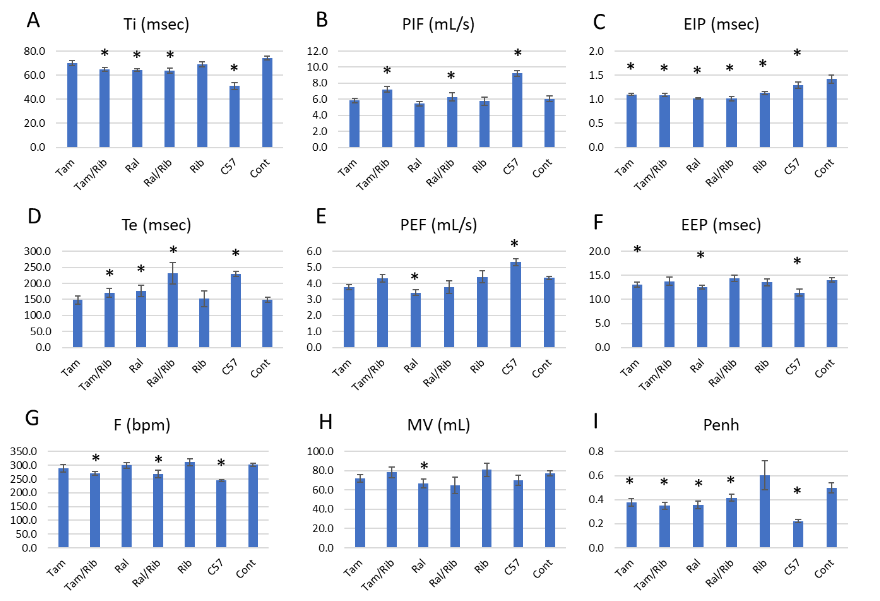
4. Plethysmography and ultrasound of P448L mice 12 months post treatment.
The progressive replacement of muscle with fibrotic tissue in the diaphragm results in significant dysfunction in respiratory parameters of the untreated P448L mouse. Using whole-body plethysmography, a number of respiratory parameters were measured to assess changes in respiration after treatment. Inspiratory (Ti) and expiratory (Te) times are increased and decreased, respectively, in the P448L mouse compared to C57 controls. While there is no change in Ti in the tamoxifen and ribitol treated groups a significant reduction was seen in all other treatment groups, though not reaching C57 levels. No apparent change was noted in the Te of tamoxifen and ribitol groups compared to controls, a significant increase however was seen in the tamoxifen/ribitol combination and raloxifene groups, reaching a similar level to C57 in the raloxifene/ribitol group. Other respiratory parameters significantly affected in the P448L mouse is a reduction in the frequency of breath (F) and increase in peak inspiratory (PIF) which were improved with the two combination groups towards C57 control. The end inspiratory pause (EIP) is consistently longer in control P448L mice compared to C57. All treatments groups show significant reduction in EIP with levels at or below C57, and the largest decrease in the raloxifene groups compared to the controls. Interestingly, positive changes in end expiratory pause (EEP) is only detected with each of the SERM treatment, but not with combined groups. Changes in PEF and MV was most obvious in the groups of raloxifene treatment, but significance is only detected with the raloxifene alone. This could be related to the body weight which is lower in the raloxifene alone group than in the combination group. Respiratory frequency (F) is normalized towards that in C57 mice in all group except for ribitol treatment alone with significance only in the two combination therapies when compared to the control P448L mice. Interestingly, enhanced pause (Penh) is significantly higher in the P448L mutant mice when compared to the C57. All the SERM treated groups showed significant decrease whereas ribitol alone treatment increases the Penh although without significance.
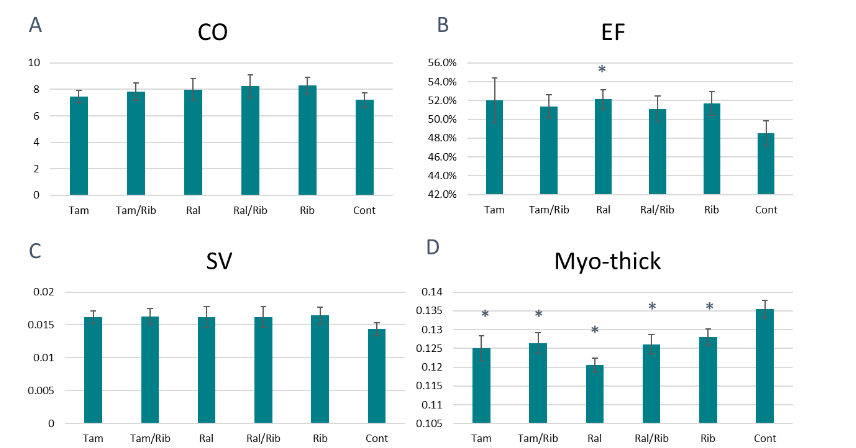
Cardiac function and morphology were also examined using high-frequency ultrasound. The P448L mutant mouse shows a reduced ejection fraction (EF) and stroke volume (SV) as well as an increase in myocardial thickness compared to C57 controls. This leads to a generally reduced overall cardiac output (CO). Treatment with tamoxifen and raloxifene, with and without ribitol, results in an increased EF of >50% compared to untreated controls (just over 48%) with statistical significance for the raloxifene alone group. Furthermore, an increased SV is noted across all treatment groups, though statistical significance was not reached. However, myocardial thickness is significantly reduced across all treatment groups with the biggest reduction noted in the raloxifene alone group. These changes result in an improved CO in all treatment groups with combined treatments showing CO levels above those of drug alone.
Discussion
Clinical trials are currently underway for the use of tamoxifen for ALS (NCT01257581) and X-linked myotubular myopathy (NCT04915846), among other disorders. The use of tamoxifen for DMD was first examined by Dorchies et al in mdx mice. A moderate dose of tamoxifen, 10 mg/kg/day, was reported to improve muscle pathology and function with reductions in fibrosis. Results from a 3-year clinical trial (NCT02835079) reported maintenance of muscle functions. A phase III clinical trial examined the effect of tamoxifen in two groups of DMD patients, aged 6.5 to 12 years with the ability to walk and under stable treatment with glucocorticoids and aged 10 to 16 years but unable to walk and not under glucocorticoid treatment (NCT03354039). Initial report published recently indicates that Tamoxifen at 20 milligrams (mg) once daily for 48 weeks is safe and well tolerated. The treatment also slows disease progression when compared to a placebo controls. However, the differences between the two groups are not clinically or statistically significant. Another recent study in vitro reported that tamoxifen treatment ameliorates contractile dysfunction of stem cell-derived cardiomyocytes on bioengineered substrates.
Our earlier study in the P448L mutant mice shows a dose dependent effect of the treatment in both the efficacy and side effects. The lowest dose of 2 mg/kg daily produces improvements in histology and function, but still incurs detectable side effect. Daily dose of 20 mg for a life-long treatment may still risk significant side effects in male reproductive organs although this potential risk is not apparent after the 48 weeks of treatment from the abovementioned clinical trial. Furthermore, it is well acknowledged that long-term use of tamoxifen is associated with increased risk of endometrial cancer. These risks seriously deter the willingness of patients and doctors to use tamoxifen for female muscular dystrophy patients. One possible solution to overcome this barrier is to establish a minimum but effective dose which avoids risk to both male and female patients. In this study we confirmed that a dose of only 1 mg/kg of tamoxifen still shows efficacy in mouse. Applying Food and Drug Administration recommended body surface area-based dosing conversion from mouse to human, the human equivalent dose would be less than 0.1 mg/kg body weight and equal to about 5 mg/kg for a 50 kg patient. This dose could be used as guidance for further clinical trials for efficacy and side effects in clinics.
One alternative means to avoid the side effects of tamoxifen is to explore the use of other SERMs with better safety record, specifically raloxifene. Raloxifene was first developed by Eli Lilly and approved by the FDA to treat postmenopausal osteoporosis more than twenty years ago. Raloxifene as an estrogen agonist acts on bone and lipid metabolism and as an estrogen antagonist for reproductive tissues. Benefits of long-term use of raloxifene include enhancement to bone mineral density, decrease of serum concentrations of total and low-density lipoprotein cholesterol, and anti-inflammatory effect. In contrast to significant negative effect of tamoxifen on endometrium and male reproductive organs, raloxifene does not stimulate the endometrium proliferation. Raloxifene does not cause significant changes in sperm production and quality, or male reproductive performance at doses as high as 100 mg/kg/daily. Therefore, raloxifene has been used as medication for many diseases with well-established safety record. Daily doses of 60-120 mg have been widely used for postmenopausal osteoporosis, lowering the risk of invasive breast cancer in high risk population and as an adjuvant for antiviral treatment including hepatitis C and SARS-CoV-2 infection. The reported effect on cell signaling and antioxidant free-radical scavenging leads to many reported applications to cardiovascular and neurodegenerative diseases such as dementia and schizophrenia. Earlier we examined the raloxifene effect in the P448L mutant mice and reported efficacy with the low dose of 50 mg /kg body weight. Applying Food and Drug Administration recommended body surface area-based dosing conversion from mouse to human, with a factor of 12, the human equivalent dose would be 250 mg/day for a 60-kg individual which is clearly higher than the standard dose currently in use for many applications, including cancer and osteoporosis. The results from this study now demonstrate that doses as low as 10 mg/kg body weight in mouse remain effective for improving muscle pathology and function. This dose will be the human equivalent dose of 50 mg daily for a 60-kg individual, well within the doses currently in clinics for long-term treatment of diseases such as osteoporosis. While treatment with a low dose of tamoxifen and raloxifene alone did not appear to restore glycosylation of α-DG, consistent with the earlier report using a high dose of tamoxifen and raloxifene, improved glycosylation was seen with combination treatment. However, this improvement is not above ribitol treatment alone suggesting a limited contribution from tamoxifen and raloxifene to matriglycan levels in the combination treatment. Additionally, a reduction in the number of centrally nucleated fibers is generally considered a sign of therapeutic efficacy. While reduced central nucleation is shown in the TA of all treatment groups an increase in centrally nucleated fibers is noted in most of the treated diaphragms, particularly the combination groups. Since CNF mainly represents regeneration, increase in CNF in all the treated groups except for the raloxifene treatment suggest a stronger regeneration capacity in the diaphragm compared to the aged control mice which may well have diminished capacity in regeneration at more than 13 months of age. This would be consistent with the lower density of fiber number in the diaphragm of the control mice (averaging ~150 fibers per 20X image area) when compared to that in the diaphragm of the treated mice (averaging >200 fibers per 20X image area). Clearly, difference in degree of degeneration, capacity in regeneration at different ages and amount of fibrosis determine the quantity of CNF in different muscle tissues. Improvements in pathology were also demonstrated with reduced fibrosis in heart, diaphragm and TA with combination treatment showing lower fibrosis than drug alone further supporting the beneficial effects of combination treatment. The histological improvements for both drug alone and combination therapies are further validated by improvements in functional results, particularly forelimb grip strength and treadmill performance as well as various plethysmography and echocardiography measures.
Conclusion
It needs to be reminded that efficacy achieved with the use of SERMs alone has been limited for treating muscular dystrophy in both human and in animal models. While the treatments alone can improve disease pathology and muscle function, averting disease progression is unlikely achievable. Combinatorial treatment is therefore clearly desirable. Here we show that combined treatment of ribitol and SERMs produces better improvement to the diseased muscles. Importantly, raloxifene and ribitol combination results in superior efficacy when compared to that achieved by individual drugs used alone. This enhanced effect is observed in both skeletal and cardiac muscles. Furthermore, better respiratory function is also detected. The two drugs have different mechanisms of action and can be considered complementary to each other. Ribitol is a metabolite with its main effect on enhancing the glycosylation of α-DG and thus unlikely to cause any negative reactions between them. While ribitol is only applicable to the muscular dystrophy caused by FKRP mutations, we reason that the multifaceted benefits of raloxifene with limited side effect demonstrated in clinics allows the potential of its use alone and in combination with many experimental therapies for muscular dystrophies including gene therapy and oligonucleotide therapy for DMD and BMD. Furthermore, our results demonstrate, encouragingly, that the SERMs, especially raloxifene at clinically applicable dosage alone and in combination with ribitol, constitutes a highly promising treatment regime for FKRP-related muscular dystrophy. Low dose raloxifene alone could also benefit other muscular dystrophies.
Conflicts of Interest Statement
The authors have no conflicts of interest to declare.
Funding Statement
This work was supported by the Carolinas Muscular Dystrophy Research Endowment at the Atrium Health Foundation (Atrium Health Foundation grant#6375)
Acknowledgements
This work was supported by the Carolinas Muscular Dystrophy Research Endowment at the Atrium Health Foundation. The authors would like to thank the vivarium staff at the James G. Cannon Research Center at the Carolinas Medical Center for their assistance in caring for the animals.
References
- Yoshida-Moriguchi T, Campbell KP. Matriglycan: a novel polysaccharide that links dystroglycan to the basement membrane. Glycobiology. Jul 2015;25(7):702-13. doi:10.1093/glycob/cwv021
- Mendell JR, Rodino-Klapac LR, Rosales XQ, et al. Sustained alpha-sarcoglycan gene expression after gene transfer in limb-girdle muscular dystrophy, type 2D. Randomized Controlled Trial Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S. Ann Neurol. Nov 2010;68(5):629-38. doi:10.1002/ana.22251
- Qiao C, Wang CH, Zhao C, et al. Muscle and heart function restoration in a limb girdle muscular dystrophy 2I (LGMD2I) mouse model by systemic FKRP gene delivery. Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t. Mol Ther. Nov 2014;22(11):1890-9. doi:10.1038/mt.2014.141
- Vannoy CH, Leroy V, Lu QL. Dose-Dependent Effects of FKRP Gene-Replacement Therapy on Functional Rescue and Longevity in Dystrophic Mice. Mol Ther Methods Clin Dev. Dec 14 2018;11:106-120. doi:10.1016/j.omtm.2018.10.004
- Vannoy CH, Xu L, Keramaris E, Lu P, Xiao X, Lu QL. Adeno-associated virus-mediated overexpression of LARGE rescues alpha-dystroglycan function in dystrophic mice with mutations in the fukutin-related protein. Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t. Hum Gene Ther Methods. Jun 2014;25(3):187-96. doi:10.1089/hgtb.2013.151
- Wu B, Drains M, Shah SN, et al. Ribitol dose-dependently enhances matriglycan expression and improves muscle function with prolonged life span in limb girdle muscular dystrophy 2I mouse model. PLoS One. 2022;17(12):e0278482. doi:10.1371/journal.pone.0278482
- Wu B, Shah SN, Lu P, et al. Long-Term Treatment of Tamoxifen and Raloxifene Alleviates Dystrophic Phenotype and Enhances Muscle Functions of FKRP Dystroglycanopathy. Am J Pathol. Apr 2018;188(4):1069-1080. doi:10.1016/j.ajpath.2017.12.011
- Wu B, Shah SN, Lu P, et al. Glucocorticoid Steroid and Alendronate Treatment Alleviates Dystrophic Phenotype with Enhanced Functional Glycosylation of alpha-Dystroglycan in Mouse Model of Limb-Girdle Muscular Dystrophy with FKRPP448L Mutation. Am J Pathol. Jun 2016;186(6):1635-48. doi:10.1016/j.ajpath.2016.02.015
- Cataldi MP, Blaeser A, Lu P, Leroy V, Lu QL. ISPD Overexpression Enhances Ribitol-Induced Glycosylation of alpha-Dystroglycan in Dystrophic FKRP Mutant Mice. Mol Ther Methods Clin Dev. Jun 12 2020;17:271-280. doi:10.1016/j.omtm.2019.12.005
- Cataldi MP, Lu P, Blaeser A, Lu QL. Ribitol restores functionally glycosylated alpha-dystroglycan and improves muscle function in dystrophic FKRP-mutant mice. Nat Commun. Aug 27 2018;9(1):3448. doi:10.1038/s41467-018-05990-z
- Gerin I, Ury B, Breloy I, et al. ISPD produces CDP-ribitol used by FKTN and FKRP to transfer ribitol phosphate onto alpha-dystroglycan. Nat Commun. May 19 2016;7:11534. doi:10.1038/ncomms11534
- Kanagawa M, Kobayashi K, Tajiri M, et al. Identification of a Post-translational Modification with Ribitol-Phosphate and Its Defect in Muscular Dystrophy. Cell Rep. Mar 8 2016;14(9):2209-23. doi:10.1016/j.celrep.2016.02.017
- Cataldi MP, Vannoy CH, Blaeser A, et al. Improved efficacy of FKRP AAV gene therapy by combination with ribitol treatment for LGMD2I. Mol Ther. Dec 6 2023;31(12):3478-3489. doi:10.1016/j.ymthe.2023.10.022
- Rosenberg AS, Puig M, Nagaraju K, et al. Immune-mediated pathology in Duchenne muscular dystrophy. Sci Transl Med. Aug 5 2015;7(299):299rv4. doi:10.1126/scitranslmed.aaa7322
- Quattrocelli M, Zelikovich AS, Salamone IM, Fischer JA, McNally EM. Mechanisms and Clinical Applications of Glucocorticoid Steroids in Muscular Dystrophy. J Neuromuscul Dis. 2021;8(1):39-52. doi:10.3233/JND-200556
- Dorchies OM, Reutenauer-Patte J, Dahmane E, et al. The anticancer drug tamoxifen counteracts the pathology in a mouse model of duchenne muscular dystrophy. Am J Pathol. Feb 2013;182(2):485-504. doi:10.1016/j.ajpath.2012.10.018
- Gayi E, Neff LA, Massana Munoz X, et al. Tamoxifen prolongs survival and alleviates symptoms in mice with fatal X-linked myotubular myopathy. Nat Commun. Nov 19 2018;9(1):4848. doi:10.1038/s41467-018-07058-4
- Awano H, Blaeser A, Keramaris E, et al. Restoration of Functional Glycosylation of alpha-Dystroglycan in FKRP Mutant Mice Is Associated with Muscle Regeneration. Am J Pathol. Jul 2015;185(7):2025-37. doi:10.1016/j.ajpath.2015.03.017
- Blaeser A, Awano H, Wu B, Lu QL. Progressive Dystrophic Pathology in Diaphragm and Impairment of Cardiac Function in FKRP P448L Mutant Mice. PLoS One. 2016;11(10):e0164187. doi:10.1371/journal.pone.0164187
- Botti V, Menzel O, Staedler D. A state-of-the-art review of tamoxifen as a potential therapeutic for duchenne muscular dystrophy. Front Pharmacol. 2022;13:1030785. doi:10.3389/fphar.2022.1030785
- Henzi BC, Schmidt S, Nagy S, et al. Safety and efficacy of tamoxifen in boys with Duchenne muscular dystrophy (TAMDMD): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. Oct 2023;22(10):890-899. doi:10.1016/S1474-4422(23)00285-5
- Birnbaum F, Eguchi A, Pardon G, Chang ACY, Blau HM. Tamoxifen treatment ameliorates contractile dysfunction of Duchenne muscular dystrophy stem cell-derived cardiomyocytes on bioengineered substrates. NPJ Regen Med. Mar 18 2022;7(1):19. doi:10.1038/s41536-022-00214-x
- Vogel VG, Costantino JP, Wickerham DL, et al. Effects of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other disease outcomes: the NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 trial. JAMA. Jun 21 2006;295(23):2727-41. doi:10.1001/jama.295.23.joc60074
- DeMichele A, Troxel AB, Berlin JA, et al. Impact of raloxifene or tamoxifen use on endometrial cancer risk: a population-based case-control study. J Clin Oncol. Sep 1 2008;26(25):4151-9. doi:10.1200/JCO.2007.14.0921
- Smith MR, Fallon MA, Lee H, Finkelstein JS. Raloxifene to prevent gonadotropin-releasing hormone agonist-induced bone loss in men with prostate cancer: a randomized controlled trial. J Clin Endocrinol Metab. Aug 2004;89(8):3841-6. doi:10.1210/jc.2003-032058
- Delmas PD, Bjarnason NH, Mitlak BH, et al. Effects of raloxifene on bone mineral density, serum cholesterol concentrations, and uterine endometrium in postmenopausal women. N Engl J Med. Dec 4 1997;337(23):1641-7. doi:10.1056/NEJM199712043372301
- Hoyt JA, Fisher LF, Buelke-Sam JL, Francis PC. The selective estrogen receptor modulator, raloxifene: reproductive assessments following premating exposure in female rats. Reprod Toxicol. May-Jun 1998;12(3):233-45. doi:10.1016/s0890-6238(98)00005-7
- Murakami Y, Fukasawa M, Kaneko Y, Suzuki T, Wakita T, Fukazawa H. Selective estrogen receptor modulators inhibit hepatitis C virus infection at multiple steps of the virus life cycle. Microbes Infect. Jan 2013;15(1):45-55. doi:10.1016/j.micinf.2012.10.003
- Montoya MC, Krysan DJ. Repurposing Estrogen Receptor Antagonists for the Treatment of Infectious Disease. mBio. Dec 18 2018;9(6) doi:10.1128/mBio.02272-18
- Peretz J, Pekosz A, Lane AP, Klein SL. Estrogenic compounds reduce influenza A virus replication in primary human nasal epithelial cells derived from female, but not male, donors. Am J Physiol Lung Cell Mol Physiol. Mar 1 2016;310(5):L415-25. doi:10.1152/ajplung.00398.2015
- Takeda M, Ikeda M, Mori K, et al. Raloxifene inhibits hepatitis C virus infection and replication. FEBS Open Bio. 2012;2:279-83. doi:10.1016/j.fob.2012.08.003
- Weickert TW, Weinberg D, Lenroot R, et al. Adjunctive raloxifene treatment improves attention and memory in men and women with schizophrenia. Mol Psychiatry. Jun 2015;20(6):685-94. doi:10.1038/mp.2015.11
- Chan YM, Keramaris-Vrantsis E, Lidov HG, et al. Fukutin-related protein is essential for mouse muscle, brain and eye development and mutation recapitulates the wide clinical spectrums of dystroglycanopathies. Hum Mol Genet. Oct 15 2010;19(20):3995-4006. doi:ddq314 [pii] 10.1093/hmg/ddq314