

Missegregation and Daily Cancer Risks: Key Insights
Missegregation Causes Potential Cancers Millions of Times every Day
Kjeld C. Engvild1
- DTU Environment, Technical University of Denmark, Roskilde, Denmark; [email protected]
OPEN ACCESS
PUBLISHED: 30 September 2025
CITATION: Engvild, KC., 2025. Missegregation Causes Potential Cancers Millions of Times every Day. Medical Research Archives, [online] 13(9). https://doi.org/10.18103/mra.v13i9.6919
COPYRIGHT: © 2025 European Society of Medicine. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
DOI https://doi.org/10.18103/mra.v13i9.6919
ISSN 2375-1924
ABSTRACT
People ask: Why do we get cancer? But perhaps it would be more pertinent to ask: Why do we not get cancer? Cells mis-segregate in about 1 percent of divisions, which means that you have a beginning of aneuploidy or potential cancer millions of times every day. If a trisomic cell (a typical result of missegregation) is allowed to divide the progeny becomes susceptible to the highly mutagenic effects of breakage-fusion-bridge cycles or to chromothripsis, where the surplus chromosome is shattered and the pieces inserted at random in the corresponding chromosome, but also elsewhere in the genome. The organism has a number of mitigating mechanisms to prevent that run-away mis-segregation/chromosome instability turns into cancer. The first line of prevention is a roll-back of the mis-segregation itself, and the cell becomes tetraploid. A second line of prevention is cell division arrest. A third line of prevention is the cellular suicide: apoptosis. A further prevention mechanism is elimination by the immune system. If all prevention mechanisms are circumvented in a cell line the result could be cancer. Large, long lived animals rarely get cancer (Peto’s paradox); they seem to have many more genes for prevention mechanisms.
Keywords: chromothripsis; malsegregation; aneuploidy; nondisjunction; endogenous cancer prevention; Warburg effect; cause of cancer; Peto’s paradox; Hansemann-Boveri
1. Introduction
Usually, people ask why do we get cancer? Lay people and physicians have wondered about this for centuries, and billions of dollars have been spent on research to find answers to this question with the hope for better cures or ways of prevention. But perhaps it might be useful to turn the problem upside down. Why do we not get cancer? Perhaps we are on the verge of getting cancer all the time, and we only avoid it because the organism has several mechanisms in place to prevent that beginning chromosome instability after missegregation turns into cancer.
It has been known for more than a hundred years, since the work of Hansemann and Boveri, that cancers most often are aneuploid and have a faulty number of chromosomes, usually many more than the normal diploid. Hansemann found grossly abnormal nuclei in cancers and Boveri observed that anomalous chromosome numbers led to cancer-like growth in fertilized sea urchin eggs. Their faulty cell division/chromosome cancer theory met little accept among their contemporaries. The aneuploidy theory of cancer has been revived by Duesberg and associates. Today it is generally agreed that cancer is a genetic disease, probably caused by mutations in 3-6 genes in a cell line; exactly how this happens has remained a major mystery.
The explanation may be that cell division in eucaryotic organisms is an error-prone process; missegregation happens all the time. Missegregation may happen in several ways: by a chromosome not being attached to the centrosome, by nondisjunction, by a lagging chromosome, by multiple centrosomes, and several others. The immediate result of missegregation is mutation in gene dosage of all the genes on a missegregated chromosome. The unfortunate consequences of this are well recognized in children with anomalous chromosome numbers.
There has been much discussion about whether the aneuploidy is a cause or a consequence of cancer, but only few oncologists have been interested in how it all starts, how aneuploidy and chromosome instability begin. The situation is different in genetic toxicology: the influence of the environment and other circumstances on carcinogenesis. There the implicit assumption is that cancer is caused by disturbance of normal chromosome segregation or DNA synthesis. An important method in genetic toxicology is the cytokinesis block micronucleus assay, where the number of chromosome missegregations are scored. In this assay lymphocytes are induced to divide with phytohemagglutinin in the presence of the possible carcinogen in question. The division is halted by cytochalasin a. When a division has gone wrong the result is a cell with two nuclei and a micronucleus with an extra chromosome. In the micronucleus assay one can see the possible carcinogenic effect of ionizing radiation and chemical mutagens, as well as the effects of living circumstances. The influence of age is almost as dramatic as the effect of carcinogens. The number of missegregations vary from below 0.1 percent in young children to almost 4 percent in old people. The estimation of chromosome missegregation is most reproducible in lymphocytes in culture, but missegregation is seen in many tissues of dividing cells such as buccal cells. In differentiated tissues such as the nervous system and liver missegregation is very common, but only of minor consequence for survival. In the rapidly dividing cells of embryos missegregation is common, but quickly corrected by apoptosis of aneuploids. In tissues like the skin, bone marrow and gut epithelium whose function is to generate new cells, run-away missegregation is very dangerous.
2. Chromosome instability after missegregation
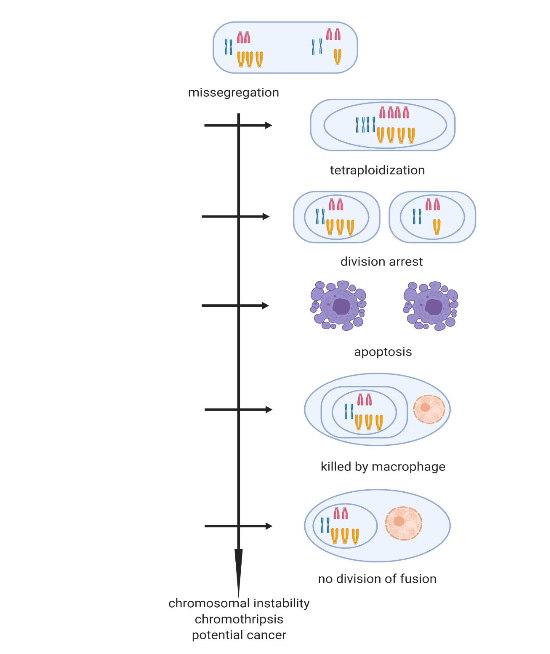
Usually one talks about chromosome instability in established cancers. However, chromosome instability begins very early in cancer development. When a standard cell division has gone wrong normal cell division is no longer possible. If a monosomic or a trisomic cell is allowed to divide, at least one of the daughter cells will have an abnormal chromosome number. In trisomic cells with a surplus chromosome there is incongruity between the cell division apparatus and the number of chromosomes. Daughter cells may end up with two extra chromosomes. With more divisions the chromosome number may increase further. This could well be a first stage of chromosome instability. It would constitute mutation of gene dosage of a whole number of genes which could well have serious consequences by itself. But there are further dangers. A trisomic cell will often have the extra chromosome sitting in a micronucleus. At division this chromosome tends to shatter and the pieces get incorporated at random, usually into the analogue chromosomes, but also into other places of the genome. This phenomenon is called chromothripsis. Cell divisions in trisomic cells may thus give rise to numerous mutations. The number of mutations may well increase with further divisions. This could well be the beginning stages of cancer.
3. A Billion potential Cancers a day
With about 300 billion cell divisions every day there are perhaps more than a billion missegregation events every day in the elderly. Each of these is a potential cancer. It has been shown that the number of cancers is proportional to the number of normal cell divisions. The organism keeps constant vigilance to prevent that these billions of potential cancers turn into real cancer. There is a whole series of mitigating mechanisms; some of these set in already before a faulty cell division is finished.
4. Mitigation mechanisms
Division roll-back. A missegregating cell may be detected by surveillance mechanisms, for example when a beginning cleavage furrow meets a lagging chromosome. The division is halted, and the result may be a binucleate cell or a cell with a tetraploid nucleus that holds two sets of chromosomes. In some cases, the result may be a cell with two diploid nuclei and a micronucleus with an encapsulated lagging chromosome.
Micronuclei elimination. If a cell division results in missegregation, the formation of micronuclei is prevented by anaphase surveillance mechanisms, thereby preventing the deleterious results of chromothripsis.
Nucleus elimination. During the formation of erythrocytes, the nucleus condenses and is expelled from the cell. The primary function of this is considered to make room for hemoglobin. Perhaps the function is to get rid of the bulky nucleus, so that small erythrocytes pass more easily through narrow capillaries. However, missegregations are not possible, when there are no nuclei and the cells cannot divide, so a function might well be prevention of missegregation.
Cell cycle arrest. Cell division is usually under strict control and differentiated cells do not normally divide. Cell cycle arrest can be induced by contact inhibition. Contact inhibition is governed by several pathways involving e.g. E-cadherin, the Hippo pathway, and a cyclin-Cdk inhibitor. Cells in dividing tissues reaching the Hayflick limit of about 50 divisions stop dividing and go into senescence; they may live for years, but do not divide. Cells with DNA damage may go into cell cycle arrest governed by the p53 complex.
Apoptosis. Damaged cells usually undergo the programmed cell death called apoptosis. Apoptosis is governed via the mitochondria and is initiated by release of proteins such as cytochrome C into the cell cytoplasm. Damage could be viral infection, nutrient stress, heat, increased calcium, or DNA faults. Apoptosis is vital for the inactivation of precancerous aneuploids and cancer cells usually harbor mutations circumventing a normal apoptosis response.
Elimination by the immune system. Missegregated aneuploid cells may be identified by the immune system and eliminated by natural killer cells. There has been much discussion about innate immune recognition of cancer and whether it might be possible to mobilize innate immune recognition in cancer treatment.
Apoptosis of macrophage fusions? Fusions of cancer cells with macrophages have been suggested as a route to metastasizing cells. A hypothetical metastasis prevention might be retention of normal apoptosis of such cell fusions.
5. Peto’s paradox
Large or long-living animals seem to have extra cancer prevention mechanisms, thus explaining that large animals with millions more cells and cell divisions than the mouse only rarely get cancer (Peto’s paradox). The mechanisms seem to be different in different species. Elephants have an extra apoptosis inducing gene, the big rodent the capybara has increased T-cell-mediated tumor suppression, and whales have a thousand tumor suppressor genes of many types. The long-lived naked mole-rat is hypersensitive to contact inhibition of cell division.
6. The p53 Dichotomy
The p53 tumor suppressor gene holds a crossroad position, central to both induction of apoptosis, cell cycle arrest and cell senescence. Many stresses induce apoptosis, but in the present context the important stressor is DNA damage. In some cases of DNA damage the p53 retards cell division, while the damage is repaired. In the case of missing telomeres the p53 induces cell senescence. In other cases DNA damage causes apoptosis. To my knowledge is it not known if aneuploidy after missegregation qualify as DNA damage. In the rapidly dividing cells of the embryo aneuploid cells are promptly eliminated by apoptosis. In the nervous system aneuploidy is the rule rather than the exception, and these cells may function normally for a hundred years. Why don’t aneuploid differentiated cells apoptose?
7. Discussion
Increased missegregations lead to aneuploidy and cancer. A lack of the spindle assembly checkpoint protein MAD1 caused 30-40% aneuploid blood cells in a 36 year old patient who contracted 12 different cancers.
Why do we get cancer? Answers have been elusive. Perhaps a more productive question could be: why do we not get cancer? The answer to this is that the organism is constantly working to keep common chromosome missegregations from turning into cancer. Only when a major part of the mitigating mechanisms has been circumvented, the result is cancer.
The most influential theories of cancer can be seen as parts of this picture. The aneuploidy theories of Hansemann and Boveri revived by Duesberg et al. describe the situation after run-away missegregation. The aerobic glycolysis theory of Warburg fits with apoptosis usually being regulated from the mitochondria; mitochondria do not function in a normal manner in cancer cells. The two-hit mutation theory of Knudson points out that several mechanisms need to be incapacitated for cancer to occur. Agents in the environment, tobacco, radiation, viruses, asbestos, etc. would in many instances interfere with one or more of the prevention mechanisms, thus explaining the cancer causes of genetic toxicology. The hallmarks of cancer describe in effect the disarming of the mitigating mechanisms.
At present it is not known which mitigation mechanism is the most important; one might suspect that apoptosis is central, and that cell cycle arrest comes second. The two are connected via the p53 tumor suppressor gene; nobody knows how many trisomic cells have entered cell cycle arrest and are sitting like small time bombs waiting for a very strong cell division signal. The elimination by the immune system would probably only apply to aneuploid cells further advanced in chromosome instability.
The existence of the many prevention mechanisms also suggests that cancer should be treated in several different ways. Aside from surgery, most cancer treatments target DNA and cell division in a broad sense. When the cancer cells die upon treatment it is usually by apoptosis or other regulated cell death. It has several times been proposed to target the apoptosis mechanism directly e.g. by using natural compounds like methyl jasmonate, menadione, betulinic acid, or resveratrol. The aerobic glycolysis typical of cancer cells (the Warburg effect) can be reversed by dichloroacetate. Dichloroacetate inhibits the formation of lactate and normalizes the citric acid cycle which might reactivate the normal apoptosis mechanism in the mitochondria. There are hundreds of papers on the effect of dichloroacetate on cancer over almost 20 years and anecdotal examples of long-term stabilization, but no phase 2 or phase 3 investigations on a potential positive palliative effect on metastatic cancer. Dichloroacetate is an approved drug used for treating lactic acidosis and it has a quite benign side effect profile (except for lung cancer patients), but it cannot be patented, so large-scale investigations can only be done with money from government or other public source.
8. Conclusions
In the long run everybody will contract cancer, when dividing cells reach the Hayflick limit. Environmental causes such as smoking, radiation, sunshine, mutagenic chemicals can only explain a certain part of cancers. It will not be possible to eliminate cancer completely by removing all environmental stressors.
Funding:
This research received no external funding.
Conflicts of Interest:
The author declares no conflict of interest.
References:
- Bignold LP, Coghlan BLD, Jersmann HPA. Hansemann, Boveri, chromosomes and the gametogenesis-related theories of tumours. Cell Biol Int 2006;30:640-644. doi:10.1016/j.cellbi.2006.04.002
- Balmain A. Cancer genetics: from Boveri and Mendel to microarrays. Nature Rev Cancer 2001;1:77-82.
- Holland AJ, Cleveland DW. Boveri revisited: Chromosome instability, aneuploidy and tumorigenesis. Nature Rev Mol Cell Biol 2009;10:478-487. doi:10.1038/nrm2718
- Duesberg P, Li RH, Fabarius A, Hehlmann R. The chromosomal basis of cancer. Cell Oncol 2005;27:293-318.
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000;100:57-70.
- Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell 2011;144:646-674. doi:10.1016/j.cell.2011.02.013
- Nordling CD. A new theory on the cancer-inducing mechanism. Brit J Canc 1953;7:68-72.
- Knudson AG. Mutation and cancer: Statistical study of retinoblastoma. Proc Nat Acad Sci 1971;68:820-823.
- Knudson, A.G. Two genetic hits (more or less) to cancer. Nature Rev Canc 2001;1:157-162.
- Tomasetti C, Marchionni L, Nowak MA, Parmigiani G, Vogelstein B. Only three driver mutations are required for the development of lung and colorectal cancers. Proc Natl Acad Sci 2015;112:118-123. doi:10.1073/pnas.1421839112
- Levine MS, Holland AJ. The impact of mitotic errors on cell proliferation and tumorigenesis. Genes Dev 2018;32:620-638. doi:10.1101/gad.314351
- Siegel JJ, Amon A. New insights into the troubles of aneuploidy. Annu Rev Cell Dev Biol 2012;28:189-214. doi:10.1146/annurev-cellbio-101011-155807
- Iemura K, Yoshizaki Y, Kuniyasu K, Tanaka K. Attenuated chromosome oscillation as a cause of chromosomal instability in cancer cells. Cancers 2021;13:4531. doi:10.3390/cancers13184531
- Bakhoum SF, Cantley LC. The multifaceted role of chromosomal instability in cancer and its microenvironment. Cell 2018;174:1347-1360. doi:10.1016/j.cell.2018.08.027
- Simonetti G, Bruno S, Padella A, Tenti E, Martinelli G. Aneuploidy: Cancer strength or vulnerability? Int J Cancer 2019;144:8-25. doi:10.1002/ijc.31718
- Fenech M. Cytokinesis-block micronucleus cytome assay. Nature Protoc 2007;2:1084-1104. doi:10.1038/nprot.2007.77
- Fenech M, Bonassi S. The effect of age, gender, diet and lifestyle on DNA damage measured using micronucleus frequency in human peripheral blood lymphocytes. Mutagenesis 2011;26:43-49. doi:10.1093/mutage/geq050
- Holland N, Bolognesi C, Kirsch-Volders M et al. The micronucleus assay in human buccal cells as a tool for biomonitoring DNA damage: The HUMN project perspective on current status and knowledge gaps. Mut Res Rev Mut Res 2008;659:93-108. doi:10.1016/j.mrrev.2008.03.007
- Luzhna L, Kathiria P, Kovalchuk O. Micronuclei in genotoxicity assessment: From genetics to epigenetics and beyond. Front Genet 2013;4:131. doi:10.3389/fgene.2013.00131
- Rehen S, Yung YC, McCreight MP et al. Constitutional aneuploidy in the normal human brain. J Neurosci 2005;25:2176-2180. doi: 10.1523/jneurosci.4560-04.2005
- Duncan AW, Newell AEH, Smith L et al. Frequent aneuploidy among normal human hepatocytes. Gastroenterology 2012;142:25-28. doi:10.1053/j.gastro.2011.10.029
- Yang M, Rito T, Metzger J et al. Depletion of aneuploid cells in human embryos and gastruloids. Nature Cell Biol 2021;23:314-321. doi:10.1038/s41556-021-00660-7
- Passerini V, Ozeri-Galai E, Pagter MS et al. The presence of extra chromosomes leads to genomic instability. Nature Commun 2016;7: art10754, 1-14. doi:10.1038/ncomms10754
- Zhang CZ, Spektor A, Cornils H et al. Chromothripsis from DNA damage in micronuclei. Nature 2015;522:179-184. doi:10.1038/nature14493
- Zhang CZ, Leibowitz ML, Pellman, D. Chromothripsis and beyond: Rapid genome evolution from complex chromosome rearrangements. Gen Devel 2013;27:2513-2530. doi:10.1101/gad.229559.113
- Voronina N, Wong JKL, Hübschmann D et al. The landscape of chromothripsis across adult cancer types. Nature Commun 2020;11: 2320. doi:10.1038/s41467-020-16134-7
- Sender R, Fuchs S, Milo, R. Revised estimates for the number of human and bacteria cells in the body. PLOS Biol 2016;14:e1002533. doi:10.1371/journal.pbio.1002533
- Sender R, Milo R. The distribution of cellular turnover in the human body. Nature Med 2021;27: 45-48. doi:10.1038/s41591-020-01182-9
- Tomasetti C, Vogelstein B. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015;347:78-81. doi:10.1126/science.1260825
- Shi QH, King RW. Chromosome nondisjunction yields tetraploid rather than aneuploid cells in human cell lines. Nature 2005;437:1038-1042. doi:10.1038/nature03958
- Orr B, Sousa F, Gomes AM et al. An anaphase surveillance mechanism prevents micronuclei formation from frequent chromosome segregation errors. Cell Reports 2021;37:109783. doi:10.1016/j.celrep.2021.109783
- Gumbiner BM, Kim NG. The Hippo-YAP signaling pathway and contact inhibition of growth. J Cell Sci 2014;127:709-717. doi:10.1242/jcs.140103
- Zhao B, Wei XM, Li WQ et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Development 2007;21:2747-2761. doi:10.1101/gad.1602907
- Mendonsa A, Na TY. Gumbiner BM. E-cadherin in contact inhibition and cancer. Oncogene 2018;37:4769-4780. doi:10.1038/s41388-018-0304-2
- Polyak K, Kato JY, Solomon MJ et al. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-β and contact inhibition to cell cycle arrest. Genes Development 1994;8:9-22.
- Shay JW. Wright WE. Hayflick, his limit, and cellular ageing. Nature Rev Mol Cell Biol 2000;1:72-76.
- Campisi J, Adda di Fagagna F. Cellular senescence: When bad things happen to good cells. Nature Rev Mol Cell Biol 2007;8:729-740. doi:10.1038/nrm2233
- Engeland, K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Diff 2018;25:114-132. doi:10.1038/cdd.2017.172
- Zhivotovsky B, Kroemer G. Apoptosis and genomic instability. Nature Rev Mol Cell Biol 2004;5:752-762. doi:10.1038/nrm1443
- Lopez J, Tait SWG. Mitochondrial apoptosis: killing cancer using the enemy within. Brit J Cancer 2015;112:957-962. doi:10.1038/bjc.2015.85
- Portt L, Norman G, Clapp C, Greenwood M, Greenwood MT. Anti-apoptosis and cell survival: a review. Biochim Biophys Acta 2011;1813:238-259. doi:10.1016/j.bbamcr.2010.10.010
- Santaguida S, Richardson A, Iyer DR et al. Chromosome mis-segregation generates cell-cycle-arrested cells with complex karyotypes that are eliminated by the immune system. Dev Cell 2017;41:638-651. doi:10.1016/j.devcel.2017.05.022
- Wang RW, Vigano S, Ben-David U, Amon A, Santaguida S. Aneuploid senescent cells activate NF-kB to promote their immune clearance by NK cells. EMBO Reports 2021;22: e52032, 1-16. doi:10.15252/embr.202052032
- Zhang HM, Chen JB. Current status and future directions of cancer immunotherapy. J Canc 2018;9:1773-1781. doi:10.7150/jca.24577
- Lu X, Kang YB. Cell fusion as a hidden force in tumor progression. Canc Res 2009;69:8536-8539. doi:10.1158/0008-5472.can-09-2159
- Shabo I, Svanvik J, Lindström A et al. Roles of cell fusion, hybridization and polyploid cell formation in cancer metastasis. World J Clin Oncol 2020;11:121-135. doi:10.5306/wjco.v11.i3.121
- Peto R. Quantitative implications of the approximate irrelevance of mammalian body size and lifespan to lifelong cancer risk. Philos Trans R Soc Lond B Biol Sci 2015;370:20150198. doi:10.1098/rstb.2015.0198
- Schiffman J, Maley CC, Nunney L, Hochberg M, Breen M (Eds.). Theme issue: Cancer across life: Peto’s paradox and the promise of comparative oncology. Philos Trans R Soc Lond B Biol Sci 2015;370.
- Vazquez JM, Sulak M, Chigurupati S, Lynch VJ. A zombie LIF gene in elephants is upregulated by TP53 to induce apoptosis in response to DNA damage. Cell Rep. 2018;24:1765-1776. doi:10.1016/j.celrep.2018.07.042
- Tejeda-Martinez D, Magalhaes JP, Opazo JC. Positive selection and gene duplications in tumour suppressor genes reveal clues about how cetaceans resist cancer. Proc R Soc B 2021;288:20202592. doi:10.1098/rspb.2020.2592
- Herrera-Alvarez S, Karlsson E, Ryder OA, Lindblad-Toh K, Crawford AJ. How to make a rodent giant: genomic basis and tradeoffs of gigantism in the capybara, the world’s largest rodent. Mol Biol Evol 2020;38:1715-1730. doi:10.1093/molbev/msaa285
- Seluanov A, Hine C, Azpurua J et al. Hypersensitivity to contact inhibition provides a clue to cancer resistance of naked mole-rat. Proc Natl Acad Sci 2009;106:19352-19357. doi:10.1073/pnas.0905252106
- Hafner A, Bulyk ML Jambhekar A, Lahav G. The multiple mechanisms that regulate p53 activity and cell fate. Nature Rev Mol Cell Biol 2019;20:199-210. doi:10.1038/s41580-019-0110-x
- Rizzotto D, Englmaier L, Villunger A. At the crossroads to cancer: How p53-induced cell fate decisions secure genome integrity. Int J Mol Sci 2021;22:10883. doi:10.3390/ijms221910883
- Villaroya-Beltri C, Osorio A, Torres-Ruiz R et al. Biallelic germline mutations in MAD1L1 induce a syndrome of aneuploidy with high tumor susceptibility. Sci Adv 2022;8: eabq5914. doi:10.1126/sciadv.abq5914
- Engvild KC. Cancer follows chromosome missegregation when all endogenous repair mechanisms fail. Med Hypot 2018;120:121-123. doi:10.1016/j.mehy.2018.08.028
- Engvild KC. Combination of the Hansemann-Boveri, Warburg, and Knudson theories of cancer, based on failure of mis-segregation damage mitigation. OBM Genetics 2019;3:art1904100. doi:10.21926/obm.genet.1904100
- Hsu P, Sabatini DM. Cancer cell metabolism: Warburg and beyond. Cell 2008;134:703-707. doi:10.1016/j.cell.2008.08.021
- Gogvadze V, Zhivotovsky B, Orrenius S. The Warburg effect and mitochondrial stability in cancer cells. Mol Asp Med 2010;31:60-74. doi:10.1016/j.mam.2009.12.004
- Blackadar CB. Historical review of the causes of cancer. World J Clin Oncol 2016;7:54-86. doi;10.5306/wjco.v7.i1.54
- Porporato PE, Filigheddu N, Pedro JMBS, Kroemer G, Galluzzi L. Mitochondrial metabolism and cancer. Cell Res 2018;28:265-280. doi:10.1038/cr.2017.155
- Michelakis ED, Webster L, Mackey JR. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Brit J Canc 2008;99:989-994. doi:10.1038/sj.bjc.6604554
- Tataranni T, Piccoli C. Dichloroacetate (DCA) and cancer: an overview towards clinical applications. Oxidative Med Cell Long 2019;art ID 8201079, 1-14. doi:10.1155/2019/8201079
- Khan A, Andrews D, Blackburn AC. Long-term stabilization of stage 4 colon cancer using sodium dichloracetate therapy. World J Clin Cases 2016;4:336-343. doi:10.12998/wjcc.v4.i10.336
- Khan, Andrews D, Shainhouse J, Blackburn AC. Long-term stabilization of metastatic melanoma with sodium dichloroacetate. World J Clin Oncol 2017;8:371-377. doi:10.5306/wjco.v8.i4.371
- Kankotia S, Stacpoole PW. Dichloroacetate and cancer: New home for an orphan drug? Biochim Biophys Acta Rev Canc 2014;1846:617-629. doi:10.1016/j.bbcan.2014.08.005
- Koltai T, Fliegel L. Dichloroacetate for cancer treatment; Some facts and many doubts. Pharmaceuticals 2024;17:744:1-60. doi:10.3390/ph17060744