

Neuro-Inflammatory Links: Autism & Lyme Disease Insights
Autism Spectrum Disorders and Lyme Disease: Exploring Shared Neuro-Inflammatory and Immune Pathways
Jodie A. Dashore¹, Brian Dashore², Scott McMahon³, Ritchie Shoemaker⁴
- Bionexus Health Clinic, Marlboro, NJ
- NYIT College of Osteopathic Medicine, Glen Head, NY
- Whole World Health Care, Roswell, NM
- ProgeneDX, Pocomoke, MD
OPEN ACCESS
PUBLISHED: 30 November 2025
CITATION: Dashore, J. A., Dashore, B., McMahon, S., Shoemaker, R., Autism Spectrum Disorders and Lyme Disease: Exploring Shared Neuro-Inflammatory and Immune Pathways. Medical Research Archives, [online] 13(11). https://doi.org/10.18103/mra.v13i11.7019
COPYRIGHT: © 2025 European Society of Medicine. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
DOI https://doi.org/10.18103/mra.v13i11.7019
ISSN 2375-1924
ABSTRACT
Building on Part 1’s exploration of Chronic Inflammatory Response Syndrome (CIRS) in pediatric neuroimmune disorders, this article (part 2 in a series of four) examines Lyme disease and its associated co-infections as infectious drivers of neuroinflammation overlapping with autism spectrum disorders (ASD). In a cohort of 1,722 children with treatment-resistant ASD and Pediatric Acute-onset Neuropsychiatric Syndrome (PANS/PANDAS), all met clinical CIRS criteria through bedside diagnosis, with notable improvements in cognition, motor skills, and gastrointestinal function following CIRS-directed therapies. Within this cohort, 47 children living in tick-endemic regions presented with Lyme-specific features including facial palsy, joint pain and swelling, difficulty chewing, cyclical fevers, and low muscle tone. They were diagnosed clinically according to CDC guidelines, supported by parental reports of bull’s-eye rashes or tick attachments. Borrelia burgdorferi and co-transmitted pathogens such as Babesia microti and Bartonella henselae can sustain immune dysregulation through mechanisms including blood–brain barrier disruption, Th17/Treg imbalance, complement activation, mitochondrial dysfunction, and microglial priming, contributing to cognitive, behavioral, and developmental impairments. Clinically, these children exhibited fatigue, attention deficits, anxiety, obsessive-compulsive behaviors, and developmental regression—features that complicate differential diagnosis with ASD. Conventional serologic testing shows limited sensitivity in early or chronic cases, underscoring the importance of clinical evaluation. While early antibiotic therapy remains the standard of care, adjunctive strategies adapted from CIRS protocols—including immune modulation and environmental remediation—appear to improve outcomes. As the second article in a four-part series on pediatric neuroimmune conditions, this work highlights infectious contributions to neurodevelopmental disruption and sets the stage for Part 3’s focus on herbal therapeutics for CIRS, the primary driver of illness in this cohort.
Introduction
As discussed in Part 1 of this series, complex neuroimmune and neuroinflammatory disorders, including Chronic Inflammatory Response Syndrome (CIRS), are increasingly recognized as contributors to chronic illness and developmental disruption in pediatric populations.¹–⁴,⁵
Part 1 highlighted how CIRS, triggered by microbial toxins and environmental exposures, overlaps with autism spectrum disorder (ASD) through shared cytokine profiles, Th17/Treg imbalances, and microglial activation, establishing a common neuroimmune endotype in some children.⁴,⁸
Among pediatric neuroimmune conditions, ASD, pediatric acute-onset neuropsychiatric syndrome (PANS) and its autoimmune subset PANDAS, and CIRS represent distinct yet often overlapping clinical entities. These disorders involve multifactorial etiologies spanning genetic susceptibility, environmental exposures, microbial triggers, immune dysregulation, and neurovascular injury—creating diagnostic and therapeutic challenges for clinicians operating within conventional frameworks.⁶,⁷
Clinicians are increasingly observing children presenting with complex symptom constellations that defy single-diagnosis models. For example, children diagnosed with autism may concurrently experience post-infectious behavioral regressions, autonomic instability, or relapsing neuropsychiatric flares—hallmarks of neuropsychiatric Lyme disease.³,⁴,⁶
Similarly, children with prior Lyme disease may develop chronic fatigue, cognitive dysfunction, and multisystem complaints indistinguishable from mold-associated illness.⁸,⁴,⁶ These observations underscore the urgent need for a systems-level understanding of how overlapping inflammatory, infectious, and immunogenetic processes coalesce into chronic pediatric syndromes.
The clinical rationale for integrating these conditions into a shared investigative and therapeutic framework is supported by mounting evidence from immunology, transcriptomics, neuroimaging, and environmental medicine. Common pathophysiological themes include persistent activation of innate immune pathways, microglial priming, blood–brain barrier compromise, mitochondrial dysfunction, and impaired detoxification mechanisms.¹,³,⁵,⁹ These mechanisms may be triggered by diverse insults—ranging from streptococcal infections to tick-borne pathogens to water-damaged building exposures—but converge on shared inflammatory networks that sustain chronic illness.
Lyme disease, caused by Borrelia burgdorferi and transmitted by Ixodes ticks, has long been recognized for its capacity to cause acute and chronic multi-system illness. In children, neuroborreliosis may present with subtle cognitive dysfunction, mood instability, and sensorimotor symptoms. Importantly, some patients continue to experience symptoms long after treatment, suggesting persistence of immune dysregulation or occult co-infections. When unresolved, post-treatment Lyme disease may trigger immune priming, mitochondrial dysfunction, and cytokine elevations that overlap with both PANS and CIRS pathophysiology.¹,³,⁸
Despite the apparent distinctions among these conditions, their co-occurrence in pediatric clinical settings is increasingly common. These children often fall through the cracks of conventional care pathways, receiving fragmented treatment for individual symptoms rather than an integrated workup of underlying inflammatory and infectious drivers. The development of a unified clinical framework—grounded in systems biology, environmental medicine, and neuroimmune science—is essential for identifying shared etiologies, stratifying risk, and implementing effective, individualized interventions.
This article presents a comprehensive scientific overview of the clinical and mechanistic intersections among autism and Lyme disease. It begins by summarizing the clinical science behind
each condition and then examines their shared immunopathological features, overlapping transcriptomic and neuroanatomical findings, and mutual exacerbating triggers. Particular attention is paid to diagnostic challenges, potential endotypes, and implications for long-term developmental outcomes. The final sections explore emerging precision medicine strategies and propose an integrated model of care for these often-misunderstood pediatric syndromes.
Methods
This is Part 2 of a four-part retrospective review series. This review builds on the clinical framework established in Part 1⁶,⁸, which investigated Chronic Inflammatory Response Syndrome (CIRS) in a cohort of 1,722 children at Bionexus Health Clinic, drawn from over 53 countries. All children were initially diagnosed with treatment-resistant autism spectrum disorder (ASD) and Pediatric Acute-onset Neuropsychiatric Syndrome (PANS/PANDAS), showing minimal improvement with standard protocols. Through detailed clinical evaluation, all met CIRS criteria, defined by multisystem symptoms including fatigue, cognitive deficits, sensory disturbances, and autonomic dysfunction, consistent with established WDB-related illness definitions⁴,⁵,⁶². PANS/PANDAS diagnoses adhered to the 2013 consensus criteria, including acute-onset obsessive-compulsive behaviors or tics⁶⁰,⁶¹. Due to unavailability of specialized laboratory tests and behavioral challenges with blood draws in ASD patients, diagnoses relied on bedside clinical assessment.
The control group, established via literature review, included 83 well children (33 from McMahon and Smith⁶², averaging 2/37 PANS symptoms; 55 from Shoemaker’s pediatric CIRS study⁴), without WDB exposure or CIRS symptoms.
Treatment with the Shoemaker protocol for CIRS, targeting biotoxin exposure and immune dysregulation, led to improvements in cognition, motor skills, gastrointestinal health, and speech/language.
In this second article, we analyzed the same cohort to explore Lyme disease and co-infections as contributors to neuroinflammatory pathology. Among the 1,722 children, 47 presented with clinical features consistent with Lyme borreliosis, including facial palsy, difficulty chewing, joint swelling and pain, difficulty walking, cyclical fevers, and low back pain. All resided in regions known to be endemic for Ixodes ticks. Parental histories provided additional context: 19 caregivers reported observing an erythema migrans (“bull’s-eye”) rash without a recalled tick bite; 7 recalled an engorged tick attached to the child; and 43 described multiple insect bites and erythema migrans–like rashes during summer months, initially presumed to be mosquito bites. According to current Centers for Disease Control and Prevention (CDC) guidance, Lyme borreliosis remains primarily a clinical diagnosis. Based on their symptomatology and exposure histories, these 47 children met bedside clinical criteria for Lyme disease and were further evaluated for potential co-infections such as Babesia and Bartonella, guided by presenting symptom patterns.
Results
The charts of 1,722 children with ASD and CIRS, previously diagnosed by established specialists, were retrospectively reviewed. There were fifty-three nationalities represented in the study group. It was noted that a subgroup of 47 children had also been diagnosed with Lyme Disease and a few with other tickborne infections. These children had some specific tickborne diseases–associated symptoms that have been listed in table. The symptoms data was compared with two separate cohorts of 55 children from a 2009 IACFS study and from a 2018 PANS/PANDAS paper presented at a recent international CIRS conference⁴,⁶².
Prior treatment for Autism consisted of recognized ASD therapies used unsuccessfully. These included specialized diets, nutritional supplements, Hyperbaric oxygen Therapy (HBOT), Fecalmicrobiota transplant (FMT), prescription anti-fungal medications, occupational therapy, speech and language therapy, feeding therapy and applied behavior analysis therapy (ABA). For the 47 children with Lyme Disease, antibiotic therapy was administered for a period of 5–14 days. 22 of the 47 children also received prescription anti-fungal medication and a probiotic supplement for the same duration as the antibiotic regimen. Parents reported no significant improvements. As per parental reports, 39 of 47 children experienced gastrointestinal distress while on the prescription medications.microbiota transplant (FMT), prescription anti-fungal medications, occupational therapy, speech and language therapy, feeding therapy and applied behavior analysis therapy (ABA). For the 47 children with Lyme Disease, antibiotic therapy was administered for a period of 5–14 days. 22 of the 47 children also received prescription anti-fungal medication and a probiotic supplement for the same duration as the antibiotic regimen. Parents reported no significant improvements. As per parental reports, 39 of 47 children experienced gastrointestinal distress while on the prescription medications.
Table 1: Autism Symptoms in the Study Group
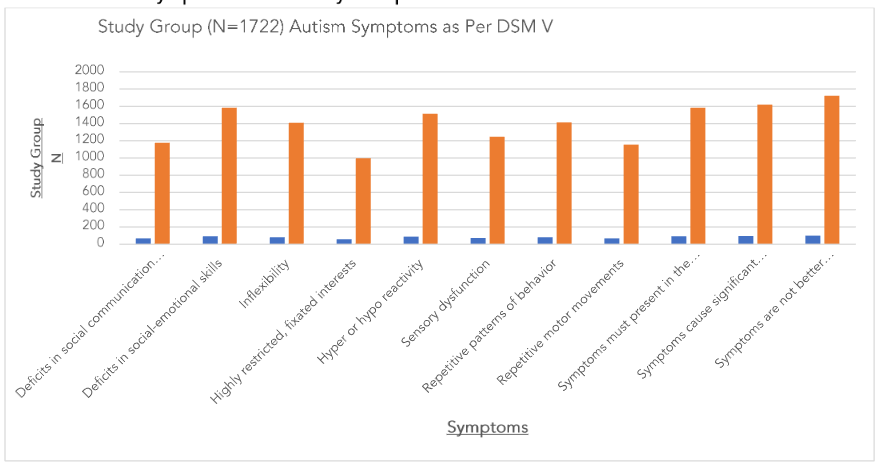
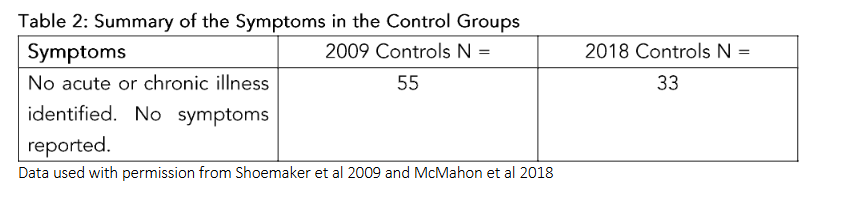
Table 3: Lyme Disease Subgroup (N = 47) Summary of Additional Symptoms Reported
| Symptoms | Pre Treatment (N =) | Percentage % | Improved Post Treatment N = | Percentage Improved % |
|---|---|---|---|---|
| Cyclical Fevers | 11 | 23% | 9 | 82% |
| Bell’s Palsy | 4 | 9% | 4 | 75% |
| Transient recurrent hemiparesis | 9 | 19% | 5 | 56% |
| Selective Mutism | 2 | 4% | 2 | 100% |
| Suicidal Ideation | 2 | 4% | 2 | 100% |
| Muscle contracture | 6 | 13% | 2 | 33% |
| Osteomyelitis (pelvis) | 1 | 2% | 1 | 100% |
| Bulls Eye Rash | 19 | 40% | 19 | 100% |
| Erythema Migrans | 43 | 91% | 32 | 74% |
| Catatonia | 1 | 2% | 1 | 100% |
| Difficulty Walking | 5 | 11% | 4 | 80% |
| Trendelenburg Gait | 7 | 15% | 4 | 57% |
| Joint pain | 47 | 100% | 41 | 87% |
| Arthritis | 14 | 30% | 11 | 79% |
| Air Hunger | 34 | 72% | 27 | 79% |
| Foot drop (+ toe walking) | 21 | 45% | 18 | 86% |
| Ptosis and visual symptoms | 19 | 40% | 11 | 58% |
| Low muscle tone | 46 | 98% | 27 | 59% |
Table 4: Summary of Coinfections Reported (N = 47)
| Coinfections | N = | % |
|---|---|---|
| Babesia | 34 | 72% |
| Anaplasma | 5 | 11% |
| Bartonella | 23 | 49% |
| Mycoplasma | 31 | 66% |
The official symptoms of ASD for the entire study cohort of 1722 cases are presented in Table 1. The data for symptoms for the control groups⁴,⁶² is shown in Table 2. The children in the control group had no acute or chronic illness and no symptoms.
Referring back to Part 1, the results The list of coinfections and associated data for this subgroup of 47 cases is highlighted in Table 4. The data for symptoms tickborne infections in the BNHC cases is presented in Table 3 along with percentages of symptoms respectively. The percentages of improvements observed per symptom are also noted in Table 3.
Looking back at the results from Part 1, treatment for CIRS utilizing the established Shoemaker methods, including alternative herbal medicine options (as highlighted in Part 3 of the series) was associated with notable improvements in cognition, motor skills, respiratory, nasal, and gastrointestinal health, along with gains reported in speech and language⁶⁸. These findings suggest that CIRS may represent a critical missing component in these treatment-resistant populations. In accordance with that, in this subgroup of 47 cases, treatment for CIRS utilizing the established Shoemaker methods, including
alternative herbal medicine options and treatment for Lyme disease utilizing botanical extracts (as highlighted in Part 3 of this series), was associated with improvements seen in associated symptoms (Table 3). These findings suggest that CIRS and Lyme Disease may represent a critical missing component in the treatment-resistant patients with ASD, and that addressing the conditions together may be considered best practice to achieve optimal clinical outcomes. Given the steadily rising prevalence of ASD, this information deserves international attention in order to assist families and provide children with respite from neuroimmune inflammatory states which can be traumatic, and impede the functional development of children with ASD.
Discussion
The findings presented in this review highlight the importance of recognizing Lyme disease and associated tick-borne infections (TBIs) as significant contributors to chronic neuroimmune dysfunction in children, particularly those diagnosed with autism spectrum disorder (ASD) or PANS/PANDAS. The methodological framework encompassed the evaluation of 1,700 children at BioNexus Health, drawn from more than 50 countries, all of whom had been classified as treatment resistant under conventional management protocols. In many of these cases, bedside clinical assessment was prioritized over laboratory confirmation, reflecting both the global unavailability of specialized diagnostic assays and the practical challenges of blood draws in children with severe behavioral or neurological impairments. Despite these limitations, a consistent clinical pattern emerged: children with overlapping neurodevelopmental and psychiatric diagnoses frequently met criteria for infection-associated chronic illness, with Lyme disease and coinfections standing out as underrecognized drivers of symptom chronicity.
This observation underscores a central theme: overlapping inflammatory cascades can obscure diagnostic clarity. Many of the children initially presented with symptom constellations attributed exclusively to ASD or PANS/PANDAS—ranging from regression and cognitive decline to anxiety, sensory disturbances, and motor dysfunction. Yet closer evaluation revealed that these manifestations were frequently sustained or amplified by tick-borne pathogens. By situating pediatric presentations within the context of environmentally acquired infection, this work reframes the etiology of persistent symptoms: rather than being explained solely by psychiatric or neurodevelopmental pathology, they often reflect an unchecked inflammatory burden driven by microbial triggers.
The improvements observed following infection-targeted interventions reinforce this perspective. Even when treatment was limited to foundational strategies—such as antibiotics, antimicrobial herbals, or supportive immunomodulation—families frequently reported sustained gains in language, behavior, gastrointestinal stability, and sleep regulation. These findings parallel broader trends in pediatric Lyme literature, where treatment-resistant cohorts demonstrate recovery once infectious and inflammatory drivers are addressed. For children with developmental vulnerabilities, such as those on the autism spectrum, these outcomes are particularly salient: they illustrate that diagnostic labels may mask underlying treatable contributors and that restoring immune balance can create conditions for developmental and functional recovery.
A methodological lesson also emerges from this synthesis. While laboratory assays for Borrelia and coinfections remain valuable, their absence did not preclude therapeutic progress. In this cohort, reliance on clinical pattern recognition allowed for timely initiation of care, validating the use of bedside criteria in resource-limited contexts. This pragmatic approach mirrors longstanding principles in infectious disease medicine, where empiric treatment is sometimes necessary to prevent prolonged morbidity. The inclusion of published control data—such as McMahon and
Smith’s cohort and Shoemaker’s pediatric analysis—further grounds these findings in comparative evidence, demonstrating that the observed trajectories are not simply coincidental but consistent with infection-mediated pathology.
The broader implication of this work is a paradigm shift in pediatric care. Conventional management of ASD or PANS/PANDAS often emphasizes psychiatric, behavioral, or immunomodulatory therapies, with little attention to the role of persistent infection. By contrast, when Lyme and coinfections are recognized as hidden drivers, initial therapeutic sequencing changes: infection-directed strategies are prioritized, creating a physiological foundation for subsequent interventions. This shift is particularly critical in children, where developmental plasticity permits meaningful recovery if inflammatory drivers are removed early.
Finally, the present findings support the integrative arc of this article series. Just as CIRS has been proposed as a concealed yet modifiable driver of chronic pediatric illness, Lyme disease and associated TBIs may represent another underappreciated factor shaping developmental and neuropsychiatric outcomes. Addressing these infections in children initially labeled as ASD or PANS/PANDAS not only broadens diagnostic precision but also expands the therapeutic window for long-term functional gains. By situating infection-driven inflammation at the forefront of clinical strategy, this work advances a systems-based approach in which pediatric neuroimmune disorders are reframed through the lens of infection, immunity, and developmental resilience.
LYME DISEASE AND ASSOCIATED CO-INFECTIONS
Lyme disease, caused primarily by Borrelia burgdorferi in North America, is the most common vector-borne illness in the United States. The infection is transmitted by Ixodes ticks, with clinical manifestations ranging from early localized erythema migrans to disseminated involvement affecting the joints, heart, and nervous system. Early recognition is often challenging because erythema migrans may be absent or atypical in up to 30–40% of cases. Untreated infection can lead to neurological complications, including meningitis, cranial neuropathies, radiculopathy, and cognitive impairment. Cardiac manifestations, such as atrioventricular conduction abnormalities, may also occur, particularly in the early disseminated phase.
Co-infections are common due to shared vectors, with pathogens such as Babesia microti and Bartonella henselae frequently co-transmitted alongside B. burgdorferi. These co-infections can amplify systemic inflammation, dysregulate immune responses, and complicate clinical presentation, often resulting in more severe fatigue, arthralgia, and neurological symptoms than Lyme disease alone. Persistent infection may stimulate chronic cytokine production, endothelial activation, and blood–brain barrier compromise, contributing to sustained inflammation and multi-organ involvement.
Clinically, Lyme disease exhibits a spectrum of manifestations influenced by host immune status, pathogen strain, and co-infections. Recognition of the complex interplay between these factors is critical for accurate diagnosis and effective management. Comprehensive assessment typically includes serologic testing, molecular diagnostics, and evaluation for potential co-infections to guide targeted therapeutic interventions and mitigate long-term sequelae.
BORRELIA BURGDORFERI: IMMUNE EVASION AND NEUROINVASION
B. burgdorferi employs sophisticated immune evasion tactics, including antigenic variation, complement inhibition, and biofilm formation. Surface proteins like VlsE undergo antigenic changes to avoid host antibody response. The bacterium also binds host complement regulators (e.g., factor H), reducing complement-mediated destruction. These strategies facilitate persistent
infection, particularly within immune-privileged sites like the central nervous system (CNS).
Neuroborreliosis occurs in approximately 10–15% of untreated cases, with presentations including meningitis, cranial neuropathies, radiculopathy, and encephalopathy²,¹⁰. B. burgdorferi can penetrate the blood–brain barrier (BBB) by modulating endothelial adhesion molecules (ICAM-1, VCAM-1), eliciting local inflammation that compromises tight junction integrity¹¹. In animal models, it has been shown to invade glial cells and perivascular neurons, activating microglia and astrocytes and increasing pro-inflammatory cytokines like IL-6, IL-17, and TNF-α¹¹,¹².
Peripheral immune activation is reflected in elevated circulating inflammatory mediators months to years after infection, particularly in patients with post-treatment Lyme disease syndrome (PTLDS)¹³. In a subset with persistent symptoms, single-photon emission computed tomography (SPECT) imaging has shown perfusion deficits in frontal and temporal lobes, correlating with cognitive impairment and mood disturbances¹⁴.
BABESIA MICROTI: PARASITIC IMMUNE DISRUPTION
Babesia microti is an intraerythrocytic protozoan that causes babesiosis, characterized by hemolytic anemia, fever, and fatigue. Co-infection with Lyme disease is well-documented; both pathogens share the Ixodes tick vector. Babesiosis can compound immune activation, exacerbate systemic cytokines, and contribute to chronic symptomatology due to persistent low-level parasitemia.
Ongoing parasitemia stimulates innate immune responses, especially macrophage activation and high levels of IFN-γ and TNF-α, which in turn can provoke anemia of inflammation and neurological symptoms¹⁵. Chronic babesiosis may perpetuate cytokine-mediated BBB dysfunction and activate microglia, although direct neuropathological studies remain limited. Clinically, concurrent infection often results in more severe fatigue, arthralgia, and cognitive fog than Lyme disease alone.
BABESIA DIAGNOSIS
Babesia infections present significant diagnostic challenges, particularly in the context of overlapping symptomatology with Lyme disease and other co-infections. Traditional blood smear evaluation remains the gold standard; Giemsa-stained thick blood film increases sensitivity for detecting intraerythrocytic parasites, though repeated sampling is often required due to fluctuating parasitemia¹⁴. Quantitative laboratory markers of hemolysis may further support diagnosis. Decreased haptoglobin levels, resulting from hemoglobin binding and clearance, provide indirect evidence of intravascular hemolysis, while the presence of free hemoglobin in urine (hemoglobinuria) reflects ongoing red blood cell destruction⁴². These findings are particularly relevant in pediatric patients, where anemia and hemolysis may manifest with fatigue, pallor, or jaundice that can be mistaken for viral or autoimmune etiologies.
Although polymerase chain reaction (PCR) assays and serology are increasingly available, their sensitivity varies with disease stage, and serologic cross-reactivity remains a confounder in endemic areas⁴². Therefore, an integrative diagnostic framework combining microscopy, biochemical markers, and molecular testing is recommended to establish Babesia infection and differentiate it from other hemolytic disorders.
CROSS-REACTIVITY OF GPI ANTIGENS
A critical challenge in the serological diagnosis of Babesia and related apicomplexan parasites is the high degree of antigenic conservation among glycosylphosphatidylinositol (GPI)-anchored molecules. These glycolipid anchors, present on the surface of Babesia, Plasmodium (malaria), Toxoplasma gondii, Eimeria, and Sarcocystis, share structural epitopes that can induce broad antibody responses⁴³. As a result, enzyme-linked immunosorbent assays (ELISAs) targeting GPI-
associated antigens often yield false-positive results, complicating interpretation in regions where multiple protozoan infections co-exist. Experimental studies have demonstrated cross-reactive IgG responses between malaria and babesiosis⁴³, and similar patterns have been observed with toxoplasmosis and coccidial infections. In clinical practice, such cross-reactivity risks overestimating prevalence rates and may lead to inappropriate treatment if relied upon without confirmatory testing. Recognition of these antigenic overlaps underscores the importance of using parasite-specific molecular assays (PCR or next-generation sequencing approaches) alongside serology, rather than depending on single-antigen ELISAs. Furthermore, deeper characterization of parasite-specific glycoconjugates is needed to refine diagnostic specificity and to develop immunoassays capable of distinguishing true Babesia infection from serologic noise caused by GPI cross-reactivity.
BARTONELLA HENSELAE: VASCULAR AND NEUROENDOCRINE EFFECTS
Bartonella spp.—notably B. henselae—are emerging as significant tick-related co-infections contributing to neurovascular and inflammatory dysfunction. The bacterium infects endothelial cells and erythrocytes, causing vasculitis, granulomatous inflammation, and increased production of VEGF and IL-8¹⁶.
Neurologically, Bartonella infection can result in encephalopathy, seizures, and neuropsychiatric symptoms such as anxiety, insomnia, and cognitive dysfunction¹⁷. In children, co-infection with Borrelia and Bartonella has been linked anecdotally to more severe neurocognitive outcomes, though large-scale quantitative studies are lacking.
ANAPLASMA PHAGOCYTOPHILUM: PATHOGENESIS AND CLINICAL MANIFESTATIONS
Anaplasma phagocytophilum is a Gram-negative, obligate intracellular bacterium transmitted by Ixodes ticks, primarily causing human granulocytic anaplasmosis (HGA)¹⁸,³⁰. The pathogen preferentially infects neutrophils, leading to functional impairment and immune dysregulation. Clinical manifestations range from mild febrile illness to severe presentations including pancytopenia, multiorgan failure, and in rare cases, encephalitis¹⁸. Laboratory findings often show leukopenia, thrombocytopenia, and elevated hepatic transaminases. Diagnosis is primarily confirmed through polymerase chain reaction (PCR) detection of bacterial DNA, while serology provides supportive evidence. Early treatment with doxycycline is highly effective; delayed therapy increases the risk of severe complications¹⁸.
Coinfections with other Ixodes-transmitted pathogens, including Borrelia burgdorferi and Babesia microti, can exacerbate the clinical course and complicate diagnosis. Emerging strains with variable virulence highlight the need for ongoing surveillance and research to optimize diagnostic accuracy and therapeutic strategies¹⁸,³⁰. HGA underscores the complex interplay between tick-borne pathogens and host immune responses, emphasizing the importance of early recognition in endemic regions.
MYCOPLASMA PNEUMONIAE: CLINICAL AND PATHOPHYSIOLOGICAL INSIGHTS
Mycoplasma pneumoniae is a cell wall–deficient bacterium that primarily infects the respiratory tract, causing atypical pneumonia and a range of extrapulmonary manifestations²². Transmission occurs via respiratory droplets, and incubation typically spans 1–4 weeks. The pathogen adheres to respiratory epithelial cells using specialized tip organelles, initiating cytotoxic effects and local immune activation²³. Infection elicits a Th1/Th17-skewed immune response, with production of proinflammatory cytokines including IL-6, IL-8, and TNF-α, contributing to pulmonary tissue damage and systemic inflammation. Extrapulmonary involvement can manifest as neurologic, cardiac, hematologic, or dermatologic complications, reflecting both direct microbial invasion and
immune-mediated processes²². Molecular mimicry between bacterial antigens and host tissues has been implicated in autoimmune phenomena post-infection. Diagnosis relies on PCR amplification of bacterial DNA or serologic detection of specific IgM and IgG antibodies, while culture is rarely used due to slow growth. Macrolides, doxycycline, or fluoroquinolones are first-line treatments, with choice guided by age, comorbidities, and local resistance patterns. Coinfection with other tick-borne pathogens, particularly in endemic regions, may exacerbate systemic inflammation and complicate clinical presentations²³. Ongoing research explores the role of persistent infection, biofilm formation, and host immune modulation in chronic or relapsing disease states. Awareness of the diverse manifestations of M. pneumoniae infection is essential for timely diagnosis and effective management, particularly in immunocompromised individuals or those with pre-existing respiratory disease.
ADDITIONAL COINFECTIONS OFTEN ASSOCIATED WITH LYME DISEASE
Ehrlichia chaffeensis is an obligate intracellular bacterium transmitted by Amblyomma ticks and is the etiologic agent of human monocytotropic ehrlichiosis (HME)¹⁹. It infects monocytes and macrophages, causing systemic immune activation and endothelial dysfunction.
Rickettsia rickettsii, the causative agent of Rocky Mountain spotted fever (RMSF), is transmitted by Dermacentor ticks and targets vascular endothelial cells, leading to systemic vasculitis and increased vascular permeability²⁰.
Tick-borne relapsing fever (TBRF) is caused by several Borrelia species, transmitted primarily through soft ticks of the genus Ornithodoros²⁴. TBRF is characterized by recurring febrile episodes separated by afebrile periods, resulting from antigenic variation of the bacterial surface proteins.
Tick-borne encephalitis virus (TBEV) is a positive-sense RNA flavivirus transmitted by Ixodes ticks, primarily Ixodes ricinus and Ixodes persulcatus, in Europe and Asia²⁶. Following a tick bite, the virus initially replicates in regional lymphoid tissues, leading to viremia and subsequent central nervous system (CNS) invasion.
MECHANISMS OF IMMUNE DYSREGULATION AND CO-INFECTION SYNERGY
Co-infections profoundly affect host immunity. Chronic exposure to Borrelia, Babesia, and Bartonella can disrupt T-cell homeostasis, skew Th1/Th17 differentiation, and suppress regulatory T-cell (Treg) function. This imbalance fosters chronic inflammation, autoantibody production, and neuroinflammatory CNS phenotypes. Similar immunomodulatory effects have been observed with other tick-borne pathogens such as Anaplasma, Ehrlichia, and Rickettsia, which can induce monocytic activation, endothelial inflammation, and cytokine cascades, further amplifying systemic immune perturbations.
Mycoplasma pneumoniae and Tick-Borne Encephalitis Virus infections also contribute via persistent antigenic stimulation, molecular mimicry, and neurotropic invasion, compounding the dysregulatory milieu. Tick-Borne Relapsing Fever spirochetes additionally promote cyclic bacteremia and recurrent immune activation, sustaining a proinflammatory environment.
Concurrently, persistent antigenic stimulation can prime microglia via peripheral-to-central immune signaling, resulting in a trained immunity state. This contributes to recurrent sensitivity to new triggers, reminiscent of PANS or post-infection encephalopathy²⁸. Endothelial activation and BBB leakage—via cytokine-mediated MMP upregulation, VEGF signaling, oxidative stress, and pathogen-induced vascular injury—facilitate immune cell infiltration and autoantibody entry into neural tissues⁶³.
Neurologically, children with Lyme and co-infections may develop regression, anxiety, mood instability, attention-deficit symptoms, OCD-like behaviors, and tics. Cognitive testing may reveal
executive dysfunction and memory impairment. These features overlap with ASD and PANS, especially in children with abrupt symptom onset or relapsing-remitting courses. The additive effects of co-infections suggest a cumulative burden on immune and neural networks, warranting a comprehensive assessment in cases with multiple tick-borne or respiratory pathogens. However, detailed clinical overlaps and biomarker patterns will be addressed in later sections.
CLINICAL PRESENTATION AND DIAGNOSTIC FRAMEWORK
Diagnosis of Lyme disease relies on clinical presentation and serology. Initial erythema migrans may be absent in up to 30–40% of cases, complicating early recognition¹. Standard two-tier serology (ELISA followed by Western blot) is recommended, though sensitivity varies—particularly in early infection or in chronic/late-stage cases. PCR testing of cerebrospinal fluid, synovial fluid, or skin biopsy can be informative, especially for neurologic involvement, but is seldom used in routine practice due to limited sensitivity²⁷.
For babesiosis, diagnosis is primarily based on blood smear visualization, PCR amplification of Babesia DNA, and serologic testing. Bartonella infections are often confirmed via blood culture or PCR, though sensitivity is low; indirect immunofluorescence assay (IFA) serology is commonly employed but demonstrates variable specificity³⁰. Other co-infections—including Anaplasma, Ehrlichia, Rickettsia, Mycoplasma pneumoniae, Tick-Borne Relapsing Fever, and Tick-Borne Encephalitis Virus—may present with nonspecific febrile syndromes, cytopenias, hepatosplenomegaly, or neurologic features, necessitating pathogen-specific molecular and serologic testing³⁰.
Given the overlapping symptomatology and high prevalence of co-infections, clinicians are encouraged to adopt a comprehensive diagnostic framework when evaluating neuropsychiatric and systemic illness associated with Lyme disease. Recommended assessments include standard and extended Lyme serologies, Babesia and Bartonella testing, pathogen-specific PCR or serology for Anaplasma/Ehrlichia/Rickettsia, complete blood count (CBC), inflammatory markers (ESR, CRP), cytokine profiling, and neuroimaging when indicated. Neurocognitive and behavioral evaluations may also reveal subtle deficits in attention, executive function, memory, and mood regulation, particularly in pediatric populations with abrupt symptom onset¹.
While detailed treatment protocols are addressed later, standard guidelines emphasize early antibiotic therapy for Lyme disease, often complemented by antimicrobial regimens targeting co-infections. Adjunctive measures—such as anti-inflammatory agents, immune modulation, and supportive care—are frequently employed in chronic cases, particularly when lingering cognitive, neuropsychiatric, or systemic symptoms persist.
Lyme disease and its co-infections represent complex multisystem disorders involving immune evasion, neurovascular compromise, and persistent systemic inflammation. These overlapping mechanisms intersect significantly with pathways implicated in autism spectrum disorder, justifying integrated neuroimmune diagnostic frameworks and mechanistic research models. While this section focuses on Lyme and co-infections, subsequent sections on Mechanistic Overlaps will further explore these intersections, emphasizing immune, endothelial, and neuroinflammatory contributions to shared phenotypes.
SHARED PATHWAYS AND DIVERGENT EXPRESSIONS
While the immune dysregulation across these disorders shares overlapping mediators, the ultimate clinical manifestation is shaped by genetic susceptibility, environmental exposures, developmental timing, and the immune milieu. For example, the same cytokine storm in a developing brain may result in regression in ASD. This
highlights the necessity of viewing these cytokine profiles as part of a broader systems-level immune network rather than isolated biomarkers.
Further, the interdependence of innate and adaptive immunity—through dendritic cell signaling, T cell polarization, and cytokine feedback loops—suggests that therapeutic interventions targeting upstream immune checkpoints may hold promise across multiple conditions. These shared immune features underscore the rationale for integrative diagnostic and therapeutic approaches grounded in immunological commonalities rather than isolated clinical silos.
NEUROVASCULAR AND BLOOD–BRAIN BARRIER DYSFUNCTION
The integrity of the neurovascular unit, particularly the blood–brain barrier (BBB), is a critical determinant of central nervous system (CNS) homeostasis. Disruption of the BBB has emerged as a unifying pathophysiological feature across autism spectrum disorder (ASD). A growing body of literature implicates vascular endothelial growth factor (VEGF), matrix metalloproteinase-9 (MMP-9), complement split product C4a, and systemic inflammation as key modulators of BBB permeability in these conditions. Understanding these mechanisms offers insight into their overlapping neuroimmune pathophysiology.
ENDOTHELIAL PERMEABILITY AND VEGF DYSREGULATION
VEGF is a potent regulator of vascular permeability and endothelial tight junction modulation. While VEGF plays an essential role in angiogenesis and neurogenesis, aberrant VEGF signaling has been implicated in pathological BBB leakage. In the context of neuroinflammation, elevated VEGF levels have been observed to disrupt endothelial tight junctions by downregulating occludin and claudin-5, key structural components of the BBB³¹. In infectious models such as neuroborreliosis, Borrelia burgdorferi has been shown to stimulate local VEGF expression in glial and endothelial cells, contributing to localized inflammation and vascular leakage¹¹.
MATRIX METALLOPROTEINASE-9 AND TIGHT JUNCTION DISRUPTION
MMP-9, a proteolytic enzyme capable of degrading components of the basal lamina and tight junctions, plays a central role in BBB disruption across multiple neuroimmune conditions. MMP-9 expression is upregulated in response to proinflammatory cytokines such as TNF-α and IL-1β and is associated with increased BBB permeability.
In Lyme disease, MMP-9 levels are also increased during acute and chronic phases, contributing to perivascular infiltration and breakdown of vascular barriers in the CNS³²,³³. Borrelia burgdorferi has been shown to induce MMP-9 secretion in astrocytes and microglia, which facilitates the organism’s transmigration across the BBB³²,³⁵. Animal models of maternal immune activation relevant to ASD have also demonstrated MMP-9 upregulation, which correlates with altered neuronal migration and synaptic patterning³²,⁵⁶.
COMPLEMENT ACTIVATION AND C4a-MEDIATED PERMEABILITY
Complement component C4a, a split product released during activation of the classical or lectin complement pathways, has emerged as a potent marker of innate immune activation in conditions such as Lyme disease, tick-borne co-infections, and autism spectrum disorders. Elevated C4a levels have been associated with endothelial injury, mast cell activation, and increased vascular permeability³⁴.
In Lyme disease, C4a is generated during innate responses to Borrelia lipoproteins, and its levels have been correlated with disease severity and vascular inflammation³⁵. Autism spectrum disorder has also been linked to dysregulated complement activity, particularly
involving early components such as C4, which are critical in synaptic refinement and CNS immune surveillance. Abnormalities in complement gene expression, including increased C4 copy number, have been proposed to contribute to aberrant neural pruning during development, potentially through microglia–endothelial signaling³⁶,³⁸.
TH17/TREG IMBALANCE AND MAST CELL ACTIVATION
T helper 17 (Th17) cells and regulatory T cells (Tregs) maintain a crucial balance in immune homeostasis. Inflammatory skewing toward Th17 dominance is a hallmark of chronic neuroimmune activation, often observed in Lyme disease. Th17 cells secrete IL-17A and IL-22, cytokines known to compromise tight junction proteins in brain endothelial cells and facilitate BBB leakage³⁷. IL-17A–mediated signaling induces the production of reactive oxygen species and activates endothelial adhesion molecules, promoting immune cell extravasation into the CNS.
Similar Th17/Treg imbalances have been identified in ASD, often accompanied by elevated proinflammatory cytokines such as IL-6 and TGF-β1, both of which influence Th17 differentiation and BBB disruption³⁸.
Mast cells are increasingly recognized as effectors of BBB modulation through release of histamine, tryptase, prostaglandins, and cytokines. In ASD, mast cell activation is frequently observed in conjunction with elevated VEGF and IL-6, which synergistically degrade endothelial integrity³⁹.
BLOOD–BRAIN BARRIER BREAKDOWN ACROSS CONDITIONS
The cumulative impact of VEGF dysregulation, MMP-9 overexpression, complement activation, and Th17 skewing results in compromised BBB architecture, allowing neurotoxic molecules, autoantibodies, and pathogens to infiltrate the brain parenchyma.
In Lyme disease, persistent infection by Borrelia species and co-infections such as Babesia and Bartonella can directly or indirectly disrupt the BBB, leading to chronic encephalopathy and neurocognitive deficits⁴⁰. These pathogens utilize adhesins and induce host enzymes such as MMPs to breach endothelial layers.
Although mechanistically diverse, these conditions converge on a final common pathway of neurovascular vulnerability. Whether driven by environmental toxins, infection, or autoimmunity, the resulting barrier dysfunction creates a permissive state for sustained neuroinflammation, maladaptive plasticity, and progressive symptomatology.
MITOCHONDRIAL DYSFUNCTION AND OXIDATIVE STRESS
Mitochondrial dysfunction and oxidative stress are increasingly recognized as core pathophysiological features across neuroimmune conditions including autism spectrum disorder (ASD)⁵⁹ and Lyme disease. These systems-level disruptions are closely intertwined, representing both upstream and downstream consequences of chronic immune activation and environmental insult. Disruptions in mitochondrial oxidative phosphorylation, excess generation of reactive oxygen species (ROS), and redox imbalance collectively drive impairments in energy metabolism, neuromodulation, and immune signaling. Mitochondrial abnormalities also intersect with neuroinflammatory signaling cascades, contributing to the phenotypic expression of fatigue, cognitive slowing, and behavioral regression.
Mitochondrial bioenergetic failure has been documented across pediatric neurodevelopmental and chronic inflammatory conditions. In ASD, multiple studies have reported impaired activities of mitochondrial electron transport chain (ETC) complexes I, III, and IV in peripheral tissues and postmortem brain. Dysfunction appears to be region-specific within the brain and is often exacerbated by immune activation or environmental triggers.
In the context of Lyme disease and co-infections, Borrelia burgdorferi and Bartonella spp. have been shown to directly interfere with host mitochondrial dynamics. Borrelia infection downregulates host mitochondrial gene expression, disrupts fission–fusion balance, and induces mitochondrial apoptosis via caspase-3 pathways¹¹. This contributes not only to neuronal loss but also to the development of central fatigue, muscle weakness, and neuropsychiatric manifestations. Furthermore, Bartonella-induced intracellular persistence may increase mitochondrial oxidative stress while impairing mitophagy, perpetuating chronic energy deficits.
Oxidative stress plays a synergistic role in damaging mitochondrial membranes and DNA. Chronic immune activation increases the generation of reactive oxygen species (ROS) and reactive nitrogen species (RNS), overwhelming endogenous antioxidant systems such as superoxide dismutase (SOD), catalase, and glutathione. Elevated levels of oxidative biomarkers, including 8-hydroxy-2′-deoxyguanosine (8-OHdG), protein carbonyls, and malondialdehyde, have been reported in ASD and Lyme disease. In children with autism, for example, increased lipid peroxidation and decreased glutathione redox ratios correlate with symptom severity and mitochondrial dysfunction, suggesting a tight link between oxidative stress and phenotypic expression.
One emerging model that integrates mitochondrial dysfunction with immune dysregulation is the concept of the “metabolic trap.” This model posits that specific metabolic intermediates, once dysregulated, can create self-reinforcing loops that perpetuate immune exhaustion and bioenergetic failure. For instance, disruption in nicotinamide adenine dinucleotide (NAD⁺) metabolism can lead to impaired sirtuin activity, altered DNA repair, and increased susceptibility to oxidative damage. Such metabolic traps have been hypothesized in ASD, wherein mitochondrial incapacity not only reduces ATP availability but also impairs immune resolution, creating a chronic inflammatory feedback loop.
Additionally, mitochondria serve as immunometabolic hubs, integrating danger signals with innate immune responses. Damaged mitochondria release danger-associated molecular patterns (DAMPs) such as mitochondrial DNA and formyl peptides, which activate pattern recognition receptors (PRRs), including toll-like receptor 9 (TLR9) and the NLRP3 inflammasome. This leads to enhanced secretion of IL-1β and IL-18, further amplifying systemic inflammation. These interactions are bidirectional; pro-inflammatory cytokines such as TNF-α and IL-6 also impair mitochondrial function via suppression of complex I activity and induction of nitric oxide synthase. In Lyme disease, such cycles may explain persistent symptoms following immune triggers despite pathogen clearance.
Neurocognitive deficits observed in Lyme and related immune-mediated disorders¹⁰,¹⁶, such as working memory impairment, processing speed delays, and executive dysfunction, are strongly linked to regional deficits in mitochondrial energy metabolism, particularly in the prefrontal cortex and hippocampus. PET imaging studies in Lyme disease have identified hypometabolism in these regions, consistent with mitochondrial energy failure.
Fatigue, a hallmark symptom of Lyme, is increasingly understood as a manifestation of systemic energy production collapse. Beyond behavioral symptoms, this fatigue is biochemically anchored in ATP depletion, redox imbalance, and impaired mitochondrial respiration. Chronic fatigue syndrome/myalgic encephalomyelitis (CFS/ME), which overlaps symptomatically with these conditions, has also been associated with similar mitochondrial deficits, supporting the shared pathomechanisms.
Finally, mitochondrial dysfunction modulates immune cell differentiation and cytokine
production. Effector T cell polarization is metabolically driven: Th17 cells rely on glycolysis and mitochondrial ROS, whereas Tregs favor oxidative phosphorylation. Perturbation of these pathways skews T cell balance toward pro-inflammatory phenotypes, exacerbating autoimmunity and delaying resolution. This immunometabolic disequilibrium is now considered a hallmark of neuroimmune disorders, linking environmental, infectious, and genetic insults with sustained mitochondrial–immune crosstalk.
MICROGLIAL PRIMING AND NEUROINFLAMMATION
Microglia are central nervous system (CNS)-resident macrophages essential for brain development, synaptic pruning, neuroplasticity, and neuroprotection³⁸. In the context of neuroimmune disorders such as Autism Spectrum Disorder (ASD)⁴¹ and Lyme disease, dysregulated microglial activity emerges as a common mechanistic thread. Persistent microglial activation—whether driven by infectious, inflammatory, or toxic insults—can result in a “primed” phenotype, wherein microglia overreact to secondary stimuli, exacerbating neuroinflammation and contributing to chronic neurological symptoms.
TRAINED IMMUNITY AND IMMUNE IMPRINTING
The concept of trained immunity—first observed in peripheral monocytes—has been extended to microglia, which display long-lasting epigenetic reprogramming after exposure to initial inflammatory triggers. Early-life immune insults⁴⁵, including maternal immune activation (MIA)⁴⁶, viral infections, or environmental toxicants, can lead to durable changes in microglial responsiveness. Such imprinting influences the developmental trajectory of neural circuits and increases the vulnerability of the CNS to subsequent immunologic or environmental challenges.
Rodent models of MIA using poly(I:C) or lipopolysaccharide (LPS) demonstrate offspring with behavioral abnormalities resembling human neurodevelopmental conditions. These effects are associated with elevated microglial expression of major histocompatibility complex class II (MHC II), interleukin-1β (IL-1β), and complement proteins such as C1q and C3. These proteins mediate aberrant synaptic pruning, a process increasingly recognized as abnormal in both ASD and schizophrenia³⁶.
In human postmortem studies, increased microglial density and activation markers such as HLA-DR and CD68 have been found in the prefrontal and cerebellar cortices of individuals with ASD. Similarly, elevated cerebrospinal fluid (CSF) levels of proinflammatory cytokines—particularly IL-6, IL-1β, and TNF-α—further support the hypothesis of chronic neuroimmune activation and microglial involvement in these conditions⁴¹.
MICROGLIAL DYSREGULATION IN ASD AND LYME DISEASE
In ASD, microglial activation is linked not only to impaired synaptic pruning but also to disrupted neurodevelopmental timing. The prefrontal cortex, amygdala, and cerebellum—all critical regions for social behavior and sensory integration—show regional microglial overactivation in ASD. This is accompanied by increased expression of oxidative enzymes like NADPH oxidase and inducible nitric oxide synthase (iNOS), both of which drive neurotoxic inflammation and mitochondrial stress.
Similarly, in Lyme neuroborreliosis, Borrelia burgdorferi can directly activate microglia via toll-like receptors (TLRs), particularly TLR2, triggering nuclear factor-kappa B (NF-κB) signaling and the release of IL-6, IL-1β, and nitric oxide¹¹. Microglial activation in this context contributes to CNS injury through excitotoxicity, increased glutamate release, and disruption of the blood–brain barrier (BBB).
MATERNAL IMMUNE ACTIVATION (MIA)
Maternal immune activation during gestation—triggered by infection, systemic inflammation, or environmental toxins—has profound implications for offspring neurodevelopment⁶⁷. Experimental models have shown that activation of the maternal immune system via viral or bacterial mimetics (e.g., poly(I:C), LPS) leads to offspring exhibiting behavioral and neuroimmune features reminiscent of ASD, schizophrenia, and anxiety disorders. Cytokines such as IL-6 and IL-17A produced during MIA can cross the placenta and disrupt fetal brain development, including cortical lamination and synaptic pruning³⁶,⁴⁵.
SYMPTOM OVERLAP BETWEEN AUTISM SPECTRUM DISORDER AND LYME DISEASE
Autism Spectrum Disorder (ASD) and Lyme disease are etiologically distinct conditions; ASD is a neurodevelopmental disorder characterized by persistent deficits in social communication and restricted, repetitive patterns of behavior, while Lyme disease is an infectious disease caused by the spirochete Borrelia burgdorferi, transmitted via Ixodes ticks. Despite these differences, clinical overlap between ASD and Lyme disease has been increasingly recognized, complicating diagnostic processes and therapeutic strategies. This section examines the neuropsychiatric, cognitive, somatic/neurological, and pediatric onset patterns shared between the two disorders, highlighting the importance of detailed clinical evaluation, careful history taking, and differential diagnosis to optimize patient outcomes.
NEUROPSYCHIATRIC OVERLAP
Neuropsychiatric manifestations are a common intersection between ASD and Lyme disease. Individuals with ASD frequently experience anxiety, irritability, mood dysregulation, and social withdrawal, which may be compounded by co-occurring conditions such as attention-deficit/hyperactivity disorder (ADHD) or sleep disturbances³⁸,⁴⁵,⁵³. Chronic Lyme disease can similarly present with depression, generalized anxiety, irritability, and emotional lability, a constellation of symptoms often described as “Lyme neuroborreliosis”⁴⁰,⁴⁶. These shared neuropsychiatric presentations suggest overlapping pathophysiological mechanisms that may include immune dysregulation, neuroinflammation, and altered neurotransmitter signaling.
Evidence indicates that Borrelia burgdorferi can cross the blood–brain barrier, triggering CNS inflammation mediated by proinflammatory cytokines such as IL-6, TNF-α, and IL-1β. This immune activation is thought to underlie the anxiety, irritability, and depression observed in chronic infection⁴⁶. Similarly, in ASD, elevated CNS levels of proinflammatory cytokines have been reported, alongside microglial activation and increased oxidative stress, suggesting a neuroimmune component to behavioral symptoms³⁸,⁵³. Shared neuroinflammatory pathways may contribute to the overlapping clinical picture, complicating differential diagnosis and potentially delaying appropriate interventions.
Clinicians should recognize that neuropsychiatric symptoms in children and adults with ASD or Lyme disease may be nonspecific and episodic, and presentation may vary over time. Comprehensive evaluation—including structured psychiatric interviews, behavioral assessments, and consideration of infection history—is critical to distinguishing between primary neurodevelopmental etiologies and infection-induced neuropsychiatric manifestations.
COGNITIVE OVERLAP
Cognitive impairments, including deficits in executive function, working memory, attention, and processing speed, are prominent in both ASD and Lyme disease. In ASD, these deficits often affect planning, problem-solving, cognitive flexibility, and social cognition, influencing academic performance and daily functioning⁴⁶.
Cognitive dysfunction in Lyme disease has been documented in both acute and chronic phases, with patients reporting memory lapses, difficulty concentrating, slowed information processing, and impaired verbal recall¹⁴,⁴⁶.
Mechanistically, cognitive impairments in both conditions may be linked to neuroinflammatory processes and mitochondrial dysfunction. In Lyme disease, neuronal injury may result from microglial activation, oxidative stress, and direct neurotoxic effects of spirochetes, whereas in ASD, mitochondrial dysfunction, oxidative stress, and neuroimmune activation have been implicated in similar cognitive deficits¹⁰,¹⁴,⁵³. This overlap highlights the need for standardized cognitive testing in individuals with either condition, allowing for quantification of deficits and monitoring of intervention efficacy.
SOMATIC/NEUROLOGICAL OVERLAP
Somatic and neurological manifestations further contribute to the diagnostic complexity between ASD and Lyme disease. Chronic fatigue, generalized weakness, headaches, and sensory sensitivities are frequently reported in both conditions. In Lyme disease, these symptoms are attributed to systemic immune activation, persistent infection, and inflammation of peripheral nerves and CNS structures¹⁰,⁴⁶. In ASD, sensory over-responsivity, gastrointestinal disturbances, and motor abnormalities may reflect underlying mitochondrial dysfunction, neuroinflammation, or comorbid medical conditions⁴.
Neurological overlap extends to fine and gross motor deficits, hypotonia, sleep disturbances, and autonomic dysregulation, including gastrointestinal dysmotility and thermoregulatory abnormalities. Both conditions demonstrate variability in symptom severity and progression, making it challenging to distinguish primary neurodevelopmental symptoms from post-infectious neurological sequelae. Clinicians should conduct thorough neurological examinations, including assessment of cranial nerve function, reflexes, coordination, and gait, in combination with laboratory markers of inflammation or infection to aid differential diagnosis.
PEDIATRIC ONSET PATTERNS & MISDIAGNOSIS RISK
The pediatric onset of ASD and Lyme disease presents a significant source of potential misdiagnosis. ASD is generally identified in early childhood, typically between ages 2 and 4, based on social communication delays, restricted interests, and repetitive behaviors. Lyme disease can affect children of all ages, with early signs including erythema migrans, fever, fatigue, and joint pain, and later stages potentially involving neurocognitive and behavioral symptoms.
Overlap in behavioral and developmental presentation can result in misdiagnosis. Children exposed to Borrelia burgdorferi may exhibit language delays, social withdrawal, irritability, and attentional difficulties that mimic core ASD features. Studies report that pediatric patients in Lyme-endemic areas are at particular risk of being diagnosed with ASD when early Lyme infection is unrecognized or untreated¹⁶,³⁸,⁴⁶. Misdiagnosis can lead to delayed treatment of the underlying infection, exacerbation of symptoms, and prolonged neurocognitive sequelae.
Early recognition requires detailed medical history, including tick exposure and prior rashes, thorough physical and neurological examinations, and targeted laboratory testing such as serologic or molecular assays for Borrelia infection. Integrating developmental assessments, behavioral evaluation, and infectious disease screening allows for accurate differentiation between ASD and infection-mediated neurodevelopmental disturbances.
TREATMENT OVERVIEW
The clinical management of complex neuroimmune syndromes such as autism spectrum disorder (ASD), Lyme disease, necessitates a multipronged approach. While treatment
strategies are often individualized, several core domains—immunological modulation, detoxification support, and symptom-targeted interventions—form the basis of comprehensive care. These conditions are rarely responsive to single-agent therapy and require dynamic protocols responsive to evolving immune, neurological, and metabolic findings.
IMMUNOMODULATION AND INFLAMMATION REDUCTION
Targeting innate and adaptive immune dysregulation is central to therapeutic approaches across these syndromes. Similarly, in Lyme-associated neuroinflammation, pulsed immunomodulatory agents are sometimes employed to reduce microglial activation and peripheral inflammatory signaling.
In ASD, particularly in those with a regressive or immune-mediated phenotype, small studies have reported benefits from interventions targeting interleukin-1β (IL-1β), tumor necrosis factor alpha (TNF-α), or mast cell stabilization³⁹,⁴⁷. However, broader clinical trials remain limited. Given the heterogeneous presentations, immune-targeted strategies are typically based on laboratory confirmation of immune activation profiles and associated symptom clusters.
DETOXIFICATION AND ENVIRONMENTAL MANAGEMENT
In patients with overlapping ASD, supportive detoxification may be considered when evidence of environmental or microbial toxicant load (e.g., mold, volatile organic compounds, lipopolysaccharide) is identified.
Nutritional interventions may support mitochondrial and antioxidant pathways, particularly in the context of reactive oxygen species (ROS) burden or metabolic blockades identified in organic acid testing or metabolomics panels. Agents such as N-acetylcysteine, coenzyme Q10, and L-carnitine have been preliminarily studied in ASD and chronic Lyme-related fatigue with variable success.
SUPPORTIVE AND INTEGRATIVE THERAPIES
Supportive interventions such as occupational and behavioral therapy, speech–language pathology, and psychotherapeutic support remain critical to long-term outcomes.
ALLOPATHIC APPROACHES: ANTIBIOTICS AND IMMUNOMODULATION
Conventional management of Lyme disease and its neuroimmune complications primarily relies on antibiotics to eradicate Borrelia burgdorferi and associated coinfections. First-line agents often include doxycycline, amoxicillin, or cefuroxime for early infection, while intravenous ceftriaxone is reserved for neurologic or cardiac involvement⁴⁸,⁴⁹. For persistent or late-stage Lyme disease, longer or repeated antibiotic courses may be employed, though evidence for post-treatment efficacy remains debated⁵⁰,⁵¹,⁵². In addition to antimicrobial therapy, anti-inflammatory and immunomodulatory agents—such as nonsteroidal anti-inflammatories, corticosteroids, or low-dose naltrexone—are sometimes used to reduce neuroinflammation and microglial activation in selected cases⁴⁹,⁵⁰. Clinical protocols are often guided by laboratory biomarkers of immune activation, co-infection status, and symptom severity, emphasizing individualized treatment plans. While antibiotics target microbial eradication, immunomodulation addresses ongoing neuroinflammatory sequelae, highlighting the need for multipronged therapy in complex or chronic presentations.
HERBAL APPROACHES:
Herbal protocols, most prominently those developed by Stephen Buhner⁵¹,⁵², offer an evidence-informed, plant-based approach to Lyme disease and coinfections. Buhner’s regimen combines multiple botanicals with antimicrobial,
References
2. Krause PJ, Narasimhan S, Wormser GP, et al. Borrelia miyamotoi sensu lato seroreactivity and seroprevalence in the northeastern United States. Emerg Infect Dis. 2014 Jul;20(7):1183-90. PMID: 24960072
3. Marques A. Chronic Lyme disease: a review. Infect Dis Clin North Am. 2008;22(2):341-360.
4. Shoemaker RC, Maizel MS. Exposure to interior environments of water-damaged buildings causes a CFS-like illness in pediatric patients: a case/control study. Bull IACFS ME. 2009;17(2):69-81.
5. Kalkbrenner AE, Schmidt RJ, Penlesky AC. Environmental chemical exposures and autism spectrum disorders: a review of the epidemiological evidence. Curr Probl Pediatr Adolesc Health Care. 2014 Nov;44(10):277-318. PMID: 25199954
6. Goyal DK, Miyan JA. Neuro-immune abnormalities in autism and their relationship with the environment: a variable insult model for autism. Front Endocrinol (Lausanne). 2014 Mar 7;5:29. PMID: 24639668
7. Brown AS. Epidemiologic studies of exposure to prenatal infection and risk of schizophrenia and autism. Dev Neurobiol. 2012 Oct;72(10):1272-6. PMID: 22488761
8. Breitschwerdt EB, Maggi RG, Nicholson WL, Cherry NA, Woods CW. Bartonella sp. bacteremia in patients with neurological and neurocognitive dysfunction. J Clin Microbiol. 2008;46(9):2856-2861.
9. Barbour AG, Fish D. Biology of Borrelia burgdorferi. Clin Microbiol Rev. 1993;6(2):127-134. PMID: 3540570
10. Halperin JJ. Nervous system Lyme disease. Infect Dis Clin North Am. 2015 Jun;29(2):241-53. PMID: 25999221
11. Ramesh G, Didier PJ, England JD, Santana-Gould L, Doyle-Meyers LA, Martin DS, Jacobs MB, Philipp MT. Inflammation in the pathogenesis of Lyme neuroborreliosis. Am J Pathol. 2015 May;185(5):1344-60. PMID: 25892509
12. Livengood JA, Gilmore RD Jr. Invasion of human neuronal and glial cells by an infectious strain of Borrelia burgdorferi. Microbes Infect. 2006 Nov-Dec;8(14-15):2832-40. Erratum in: Microbes Infect. 2015 Jun;17(6):e1. PMID: 17045505
13. Klemen Strle, Daša Stupica, Elise E. Drouin, Allen C. Steere, Franc Strle, Elevated Levels of IL-23 in a Subset of Patients With Post–Lyme Disease Symptoms Following Erythema Migrans, Clinical Infectious Diseases, Volume 58, Issue 3, 1 February 2014, Pages 372–380
14. Fallon BA, Keilp JG, Corbera KM, et al. Regional cerebral blood flow and cognitive deficits in chronic Lyme disease. J Neuropsychiatry Clin Neurosci. 2003;15(3):326-332. PMID: 12928508
15. Vannier EG, Krause PJ. Human babesiosis. N Engl J Med. 2012; 366(25):2397-2407. PMID: 22716978
16. Breitschwerdt EB, Maggi RG, Nicholson WL, Cherry NA, Woods CW. Bartonella sp. bacteremia in patients with neurological and neurocognitive dysfunction. J Clin Microbiol. 2008 Sep;46(9):2856-61. PMID: 18632903
17. Chomel BB, Kasten RW. Bartonellosis, an increasingly recognized zoonosis. J Appl Microbiol. 2010 Sep;109(3):743-50. PMID: 20148999
18. Dumic I, Jevtic D, Veselinovic M, et al. Human granulocytic anaplasmosis: a systematic review of published cases. Microorganisms. 2022;10(7):1433. PMID: 35889152
19. Paddock CD, Childs JE. Ehrlichia chaffeensis: a prototypical emerging pathogen. Clin Microbiol Rev. 2003;16(1):37-64. PMID: 12525424
20. Dantas-Torres F. Review: Rocky Mountain spotted fever. Lancet Infect Dis. 2007;7(4):201-207. PMID 17961858
21. Biggs HM, Behravesh CB. Diagnosis and management of tickborne rickettsial diseases: Rocky Mountain spotted fever, ehrlichiosis, and anaplasmosis—United States. MMWR Recomm Rep. 2016;65(2):1-44.
22. Waites KB, Talkington DF. Mycoplasma pneumoniae and its role as a human pathogen. Clin Microbiol Rev. 2017;30(2):747-809.
23. Atkinson TP, Balish MF, Waites KB. Epidemiology, clinical manifestations, pathogenesis, and laboratory detection of Mycoplasma pneumoniae infections. FEMS Microbiol Rev. 2008;32(6):956-73. PMID 18754792
24. Cutler SJ. Relapsing fever borreliae: a global review. Clin Lab Med. 2010;30(1):149-160.
25. Dworkin MS, Schwan TG, Anderson DE, Borchardt SM. Tick-borne relapsing fever. Infect Dis Clin North Am. 2008;22(3):449-68. PMID 18755384
26. Süss J. Tick-borne encephalitis 2010: epidemiology, risk areas, and virus strains in Europe and Asia—an overview. Ticks Tick Borne Dis. 2011 Mar;2(1):2-15. PMID 21771531
27. Bogović P, Lotrič-Furlan S, Avšič-Županc T, et al. Comparison of clinical, laboratory and immune characteristics of the monophasic and biphasic course of tick-borne encephalitis. Microorganisms. 2021 Apr 10;9(4):796. PMID 33920166
28. Robinson-Agramonte MLA, Noris García E, Fraga Guerra J, Vega Hurtado Y, Antonucci N, Semprún-Hernández N, Schultz S, Siniscalco D. Immune Dysregulation in Autism Spectrum Disorder: What Do We Know about It? Int J Mol Sci. 2022 Mar 11;23(6):3033. PMID: 35328471
29. Wormser GP, Dattwyler RJ, Shapiro ED, Halperin JJ, Steere AC, Klempner MS, Krause PJ, Bakken JS, Strle F, Stanek G, Bockenstedt L, Fish D, Dumler JS, Nadelman RB. The clinical assessment, treatment, and prevention of Lyme disease, human granulocytic anaplasmosis, and babesiosis: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2006 Nov 1;43(9):1089-134. Clin Infect Dis. 2007 Oct 1;45(7):941. PMID: 17029130
30. Ismail N, Bloch KC, McBride JW. Human ehrlichiosis and anaplasmosis. Clin Lab Med. 2010 Mar;30(1):261-92. PMID: 20513551
31. Argaw AT, Asp L, Zhang J, et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J Clin Invest. 2012 Jul;122(7):2454-68. PMID:
22653056
32. Perides G, Charness ME, Tanner LM, Péter O, Satz N, Steere AC, Klempner MS. Matrix metalloproteinases in the cerebrospinal fluid of patients with Lyme neuroborreliosis. J Infect Dis. 1998 Feb;177(2):401-8. PMID: 9466528
33. Szczepanski A, Furie MB, Benach JL, Lane BP, Fleit HB. Interaction between Borrelia burgdorferi and endothelium in vitro. J Clin Invest. 1990 May;85(5):1637-47. PMID: 2332509
34. Talkington J, Nickell SP. Borrelia burgdorferi spirochetes induce mast cell activation and cytokine release. Infect Immun. 1999;67(3):1107-15. PMID: 10024550
35. Brissette CA, Bykowski T, Cooley AE, Bowman A, Stevenson B. Borrelia burgdorferi RevA antigen binds host fibronectin. Infect Immun. 2009 Jul;77(7):2802-12. PMID: 19398540
36. Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, Tooley K, Presumey J, Baum M, Van Doren V, Genovese G, Rose SA, Handsaker RE; Schizophrenia Working Group of the Psychiatric Genomics Consortium; Daly MJ, Carroll MC, Stevens B, McCarroll SA. Schizophrenia risk from complex variation of complement component 4.Nature. 2016 Feb 11;530(7589):177-83. PMID: 26814963
37. Kebir H, Kreymborg K, Ifergan I, et al. Human Th17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med. 2007 Oct;13(10):1173-5. PMID: 17828272
38. Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Pessah IN, Van de Water J. Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav Immun. 2011 Jan;25(1):40-5. PMID: 20705131
39. Theoharides TC, Tsilioni I, Patel AB. Atopic diseases and inflammation of the brain in the pathogenesis of autism spectrum disorders. Transl Psychiatry. 2016 Jun;6(6):e844. PMID: 27351598
40. Halperin JJ. Nervous system Lyme disease. Infect Dis Clin North Am. 2015 Jun;29(2):241-53. PMID: 25999221
41. Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol. 2005 Jan;57(1):67-81. PMID: 15546155
42. Homer MJ, Aguilar-Delfin I, Telford SR 3rd, Krause PJ, Persing DH. Babesiosis. Clin Microbiol Rev. 2000 Jul;13(3):451-469. PMID: 10885987
43. Gowda DC. Structure and activity of glycosylphosphatidylinositol anchors of Plasmodium falciparum. Microbes Infect. 2002 Jul;4(9):983-90. PMID: 12106792
44. Shoemaker RC, Maizel MS. Exposure to interior environments of water-damaged buildings causes a CFS-like illness in pediatric patients: a case/control study. Bull IACFS ME. 2009;17(2):69-81
45. Choi GB, Yim YS, Wong H, et al. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science. 2016 Feb 26;351(6276):933-9. PMID: 26822608
46. Bransfield RC. Neuropsychiatric Lyme borreliosis: an overview with a focus on a specialty psychiatrist’s clinical practice. Healthcare (Basel). 2018 Aug 25;6(3):104. PMID: 30149626
47. Masi A, Quintana DS, Glozier N, Lloyd AR, Hickie IB, Guastella AJ. Cytokine aberrations in autism spectrum disorder: a systematic review and meta-analysis. Mol Psychiatry. 2015 Apr;20(4):440-6. PMID: 24934179
48. Steere AC, Strle F, Wormser GP, et al. Lyme borreliosis. Nat Rev Dis Primers. 2016 Dec 15;2:16090. PMID: 27976670
49. Cameron DJ, Johnson LB, Maloney EL. Evidence assessments and guideline recommendations in Lyme disease: the clinical management of Lyme disease. Expert Rev Anti Infect Ther. 2014 Sep;12(9):1103-35. PMID: 25077519
50. Marques A. Chronic Lyme disease: a review. Infect Dis Clin North Am. 2008 Jun;22(2):341-60. PMID: 18452806
51. Buhner S. Herbal Antibiotics, 2nd Edition: Natural Alternatives for Treating Drug-Resistant Bacteria. Storey Publishing; 2015.
52. Feng J, Leone J, Schweig S, Zhang Y. Evaluation of Natural and Botanical Medicines for Activity Against Growing and Non-growing Forms of B. burgdorferi. Front Med (Lausanne). 2020 Feb 21;7:6. PMID: 32154254
53. Pangrazzi, L.; Balasco, L.; Bozzi, Y. Oxidative Stress and Immune System Dysfunction in Autism Spectrum Disorders.Int. J. Mol. Sci. 2020, 21, 3293. PMID: 32384730
54. Tylee DS, Hess JL, Quinn TP, et al. Blood transcriptomic comparison of individuals with and without autism spectrum disorder: A combined-samples mega-analysis. Am J Med Genet B Neuropsychiatr Genet. 2017 Apr;174(3):181-201. PMID: 27862943
55. Aucott JN, Soloski MJ, Rebman AW, et al. CCL19 as a chemokine risk factor for posttreatment Lyme disease syndrome: a prospective clinical cohort study. Clin Vaccine Immunol. 2016 Sep 6;23(9):757-66. PMID: 27358211
56. Scott, O., Shi, D., Andriashek, D., Clark, B. and Goez, H.R. (2017), Clinical clues for autoimmunity and neuroinflammation in patients with autistic regression. Dev Med Child Neurol, 59: 947-951.PMID 28383115
57. Hamilton D. Understanding mycotoxin-induced illness: Part 1. Altern Ther Health Med. 2022 Jul;28(4):8-10. PMID: 36069791
58. Butovsky O, Weiner HL. Microglial signatures and their role in health and disease. Nat Rev Neurosci. 2018 Oct 19;19(10):622-635. PMID: 30206328
59. Frye, R.E. Mitochondrial Dysfunction in Autism Spectrum Disorder: Unique Abnormalities and Targeted Treatments. Semin. Pediatr. Neurol. 2020, 35, 100829, PMID:32892956
60. Chang K, Frankovich J, Cooperstock M, et al. Clinical evaluation of youth with pediatric acute-onset neuropsychiatric syndrome (PANS): recommendations from the 2013 PANS Consensus Conference. J Child Adolesc Psychopharmacol. 2015 Feb;25(1):3-13. PMID:25325534
61. Frankovich J, Swedo SE, Murphy TK, et al. Clinical management of pediatric acute-onset neuropsychiatric syndrome: part II—use of immunomodulatory therapies. J Child Adolesc Psychopharmacol. 2017 Sep;27(7):574-593. PMID: 36358107
62. McMahon SW, Smith J. Pediatric Quirks of CIRS and PANS. Paper presented at: Surviving Mold Annual Conference; May 4th, 2018; Salisbury, MD
63. Shoemaker RC, House DE. Sick building syndrome (SBS) and exposure to water-damaged buildings: time series study, clinical trial and mechanisms. Neurotoxicol Teratol. 2006 Sep-Oct;28(5):573-88. PMID 17010568
64. Shimasaki C, Frye RE, Trifiletti R, Cooperstock M, Kaplan G, Melamed I, et al. Evaluation of the Cunningham Panel™ in pediatric autoimmune neuropsychiatric disorder associated with streptococcal infection (PANDAS) and pediatric acute-onset neuropsychiatric syndrome (PANS): Changes in antineuronal antibody titers parallel changes in patient symptoms. J Neuroimmunol. 2020 Feb 15;339:577138. PMID:
31884258
65. Gebbia JA, Coleman JL, Benach JL. Borrelia spirochetes upregulate release and activation of matrix metalloproteinase gelatinase B (MMP-9) and collagenase 1 (MMP-1) in human cells. Infect Immun. 2001 Jan;69(1):456-62. PMID: 11119537
66. Estes ML, McAllister AK. Maternal immune activation: implications for neuropsychiatric disorders. Science. 2016 Aug 19;353(6301):772–7. PMID: 27540164
67. Zawadzka A, Cieślik M, Adamczyk A. The Role of Maternal Immune Activation in the Pathogenesis of Autism: A Review of the Evidence, Proposed Mechanisms and Implications for Treatment. International Journal of Molecular Sciences. 2021; 22(21):11516. PMID: 34768946
68. Dashore, JA., Dashore, B., et al., 2025. Chronic Inflammatory Response Syndrome: Exploring Neuroimmune Pathology and Multisystem Framework for Differential Diagnosis in Pediatrics- Part 1. Medical Research Archives, [online] 13(11).
https://doi.org/10.18103/mra.v13i11.6952
Most read articles by the same author(s)
- Jodie A. Dashore, PhD, OTD, RH (AHG), Brian Dashore, DO, Scott McMahon, MD, Ritchie Shoemaker, MD, Chronic Inflammatory Response Syndrome: Exploring Neuroimmune Pathology and Multisystem Framework for Differential Diagnosis in Pediatrics- Part 1 , Medical Research Archives: Vol 13 No 10 (2025): Vol.13, Issue 10, October 2025
Conclusion
Recognition of pediatric neuroimmune vulnerability may manifest as cognitive inflexibility, behavioral regression, sensory hypersensitivity, or motor tics prior to the emergence of more recognizable systemic symptoms. Longitudinal studies indicate that pediatric neuroimmune disorders can manifest as developmental deceleration, attentional shifts, or dysregulated arousal, often preceding laboratory-confirmed infection or inflammatory marker elevation.
Funding Statement
The authors have no conflicts of interest to declare.
Acknowledgments
No acknowledgments.
References
1. Krause PJ, Narasimhan S, Wormser GP, et al. Borrelia miyamotoi sensu lato seroprevalence and seropositivity in the northeastern United States. Emerg Infect Dis. 2014 Jul;20(7):1183-90. PMID: 24960072
2. Marques A. Chronic Lyme disease: a review. Infect Dis Clin North Am. 2008;22(2):341-360.
3. Shoemaker RC, Maizel MS. Exposure to interior environments of water-damaged buildings causes a case/control study. Bull IACFS ME. 2009;17(2):69-84.
“`