






Neurodevelopmental Insights in MPS II: A Retrospective Study
Retrospective Natural History Study of neurodevelopment in neuronopathic Mucopolysaccharidosis Type II
Dawn Phillips¹, Yoonjin Cho¹, Caroline Mulatya¹, Mark Forsberg¹, Michele Poe², Maria Escolar²
- REGENXBIO Inc., Rockville, MD, USA
- Children’s Hospital of Pittsburgh, University of Pittsburgh Medical Center, Pittsburgh, PA, USA.
OPEN ACCESS
PUBLISHED: 30 November 2024
CITATION: Phillips, D., Cho, Y., et al., 2024. Retrospective Natural History Study of neurodevelopment in neuronopathic Mucopolysaccharidosis Type II. Medical Research Archives, [online] 12(11). https://doi.org/10.18103/mra.v12i11.5915
COPYRIGHT: © 2024 European Society of Medicine. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
DOI https://doi.org/10.18103/mra.v12i11.5915
ISSN 2375-1924
Abstract
Mucopolysaccharidosis Type II (MPS II) is a X-linked lysosomal storage disease caused by deficiency of iduronate-2-sulfatase (I2S) leading to accumulation of glycosaminoglycans and in the neuronopathic form, results in irreversible neurocognitive decline. The objectives of this study were to better understand the developmental trajectories of patients with neuronopathic MPS II across multiple domains and to identify timepoints where patients deviate outside of typical development and where skill acquisition plateaus.
This study presents a retrospective non-interventional medical records review of the neurodevelopmental natural history of patients with neuronopathic MPS II using the Mullen Scales of Early Learning (MSEL) Visual Reception, Expressive Language, Receptive Language, and Fine Motor scales. Function was characterized relative to MSEL normative ±1SD and ±2SD boundaries.
MSEL natural history data was available for 32 patients. The majority of the children were on enzyme replacement therapy. Developmental trajectories deviated below –1SD from the normative mean at a mean chronological age range from 12.8 (Expressive Language) to 19.5 months (Visual Reception) and below -2SD from 24.0 (Fine Motor) to 29.3 months (Visual Reception). The mean chronological age where skill acquisition plateaued ranged from 66.1 to 74.6 months while the developmental age equivalence score where skill acquisition plateaued ranged from 25.7 to 29.9 months. Slowing in the rate of skill acquisition was present very early in development and varied by baseline function and chronological age.
Visual Reception skill acquisition when developmental function was -1SD from the normative mean was 5.8 months/year, at -2SD was 4.7 months/year and, from -2SD to peak skill acquisition, the mean rate was 1.6 months/year.
Results of this study support that developmental delay in patients with neuronopathic MPS II occurs across multiple domains very early in development and that the rate of skill acquisition varies by baseline function and chronological age. Although some children may acquire a small number of skills until they are 5–6 years old, function does not typically exceed the 2–3-year-old level and the rate of change per year is thereafter very small. Use of a multi-domain measure is important to understand disease impacts and developmental variability in neuronopathic MPS II.
Keywords
Mucopolysaccharidosis Type II, neurodevelopment, enzyme replacement therapy, Mullen Scales of Early Learning, developmental trajectories
1. Introduction
Mucopolysaccharidosis Type II (MPS II, Hunter syndrome) (OMIM #309900), is a rare, autosomal recessive, X-linked lysosomal storage disorder caused by a mutation in the IDS gene leading to a deficiency of iduronate-2-sulfatase (I2S). This results in the accumulation of glycosaminoglycans (GAGs) in various tissues causing dysfunction in multiple organ systems. GAG accumulation results in typical storage lesions and diverse disease symptoms, with the central nervous system (CNS) being minimally affected in the non-neuronopathic (attenuated) form of the disease. However, there is irreversible neurocognitive decline in patients with the neuronopathic phenotype (severe), which represent two-thirds of total patients.
In neuronopathic MPS II, GAGs accumulate in the neuronal tissue leading to progressive cognitive and behavioral deterioration, global developmental delay, epilepsy, sensorineural hearing loss, language impairment, retinal degeneration, and sleep problems. Additional systemic features include facial dysmorphism, progressive skeletal dysplasia, joint contractures, carpal tunnel syndrome, severe airway disease, cardiac valve disease and hepatosplenomegaly. Children with MPS II appear normal at birth, but in the neuronopathic form, signs, and symptoms can present as early as 7.3 months of age and natural history data indicate that skill acquisition can slow down as early as 6–9 months of age.
In the first 12 months of life, some patients fail hearing screening tests, may present with speech delay and may not meet typical developmental milestones. Neurodevelopmental function follows the course of a delay phase, followed by a plateau phase at an average age of 4 to 4.5 years and then a decline phase. Limited information is available with respect to developmental trajectories in the delay stage. The delay phase is defined as the stage where children continue to acquire skills but at a much slower rate than typically developing children and function increasingly deviates further from normal. Morbidity and mortality are high in MPS II, with death reported to occur at mean ages of 11.8 years in patients with the neuronopathic phenotype and 21.7 years in patients with a mild or attenuated phenotype.
The management of MPS II requires a multidisciplinary approach with surgical and supportive interventions helping to manage physical manifestations of the disease. Enzyme replacement therapy (ERT) may help improve systemic symptoms and survival but does not cross the blood–brain barrier and is therefore ineffective in preventing, treating, or reversing the developmental decline of neuronopathic disease.
External natural history data sets may guide understanding of disease trajectories in the natural progression of the disease and serve as comparator samples for interventional studies. The Mullen Scales of Early Learning (MSEL) has been identified as a valid and reliable outcome measure for use in MPS II clinical trials, and is widely used in children with symptoms seen in MPS II, such as behavior and attention challenges, hearing and vision loss, motor impairment, and health issues. The MSEL assesses typical motor, language, and cognition milestones and includes direct observation of typical function (e.g., talking, walking, reach, and grasp) and use of standardized manipulatives to measure developmental concepts (e.g., visual tracking, sorting, and matching objects, drawing, and building with blocks). The MSEL Visual Reception and Fine Motor scales are mostly nonverbal and the test objects and pictures are not labelled or culturally specific. The Visual Reception, Expressive and Receptive Language, Fine and Gross Motor scales provide a comprehensive assessment of development from birth to 68 months (Gross Motor until 33 months) across multiple domains. Metrics include raw, age equivalent (AEq), T-scores and an Early Learning Composite score. Age equivalent scores represent the average age in months in the normative sample in which a given total raw score is typical. T-scores describe function relative to a normative reference sample of typically developing children with a mean of 50 and a standard deviation (SD) of 10.
The objectives of this study were to better understand the developmental trajectories of patients with neuronopathic MPS II across multiple domains, to identify early timepoints where patients deviate outside of typical development, and to identify where skill acquisition plateaus. This study utilized a review of retrospective, non-interventional medical records of the neurodevelopmental natural history of patients with neuronopathic MPS II using the MSEL.
2. Methodology
2.1 RECRUITMENT
Recruitment took place at three medical centers known to diagnose and treat patients with MPS II. This manuscript will focus on the neurodevelopmental data that was available from a single medical center because the other clinical sites did not have available longitudinal data. The study protocol included a wide range of neurodevelopmental assessments, but the MSEL was the only developmental assessment with available data. All aspects of the study were managed by the Sponsor of the study REGENXBIO Inc., including oversight of the contract research organization (CRO) that provided the study center management. All patients had a confirmed diagnosis of neuronopathic MPS II either through genotype variant confirmation or established developmental delay, were male and had at least one MSEL neurodevelopmental assessment in their medical records. The subject, subject’s parent(s), or legally authorized guardian(s) voluntarily signed an IRB/IEC-approved ICF.
2.2 CHARACTERIZATION OF THE MPS II NATURAL HISTORY DISEASE TRAJECTORY RELATIVE TO TYPICALLY DEVELOPING CHILDREN
MSEL normative data were used to characterize SD for AEq for normally developing children. Mock data points were collected from the MSEL manual normative data, and an algorithm was created to map each score to discrete T-scores at ±1SD and ±2SD boundaries. Linear regression models were fitted between mapped AEq scores and age, and the slopes from the regression lines were referenced to analyze the developmental function. Modeling was used to estimate the boundary for AEq based on normative data of <68 months, so caution should be taken when extrapolating boundaries for older children (>68 months). T-scores describe developmental function relative to a normative reference sample and indicate rate of skill acquisition. If the rate of skill acquisition is slower than the normative reference sample, additional values are necessary to interpret change in function. The modeling approach allows AEq to be used to show skill acquisition or decline and provides a value that can be compared within and across developmental scales. The creation of standard deviation boundaries supports AEq use to compare to the normative sample for the MSEL and to define key timepoints where developmental function deviates outside of the normal range.
MSEL trajectories of neuronopathic MPS II were analyzed for all scales except Gross Motor because the scale can only be administered from birth through 33 months, as opposed to from birth through 68 months for the other MSEL scales. Mean AEq trajectories were plotted relative to the MSEL normative AEq SDs for each MSEL scale. The mean AEq trajectories are displayed with the following timepoints: chronological age when the AEq mean trajectory deviates below the normative mean by -1SD and -2SD and the inflection timepoint where participants reach the mean maximum AEq score before the trajectory levels off. Disease progression was further characterized by defining the mean developmental age when the trajectory deviates below the normal population by -1SD and -2SD based on a quadratic mixed model. The mean gain in AEq from -2SD below the normative mean to the inflection point is also reported as the total gain in months AEq/52 weeks of age, based on quadratic mixed modeling between chronological age and AEq. The estimated AEq score change per year was calculated for each MSEL scale from chronological age intervals of 3 months using a piecewise linear mixed model.
3. Results
3.1 PARTICIPANT CHARACTERISTICS
MSEL natural history data was available for 32 participants with neuronopathic MPS II confirmed by genotype mutation, family history or cognitive decline. Participant characteristics are shown in Table 1. The mean age of subjects at first MSEL assessment was 38.4 ± 23.3 months. The genotype mutations were available for 22 participants and included nonsense (27.3%), inversion and missense R468 (18.2%, each), and frameshift, copy number variant, inversion splicing and missense (9.1% each), as characterized in Seo et al. 2020. The majority of participants were receiving ERT and three had undergone a hematopoietic stem cell transplantation (HSCT).
| Demographics | Age at First MSEL Assessment (months) | Sex | Race | Mutation Subtype |
|---|---|---|---|---|
| N | 32 | 32 | 32 | 22 |
| Mean | 38.38 | Male 32 (100%) | Asian 3 (9.4%) | Copy number variant 2 (9.1%) |
| SD | 23.28 | Black or African American 3 (9.4%) | Other 2 (6.3%) | Inversion 4 (18.2%) |
| Median | 39.50 | Unknown 2 (6.3%) | White 22 (68.8%) | Missense 2 (9.1%) |
| (Min, Max) | (1, 117) | Missense R468 4 (18.2%) | ||
| Nonsense 6 (27.3%) | ||||
| Splicing 2 (9.1%) | ||||
| Unknown 2 (9.1%) |
3.2 MSEL T-SCORES
A decline in T-scores on some MSEL scales was evident very early in development and often apparent as early as the interval from the first to second assessment, though individual patient developmental function may be variable among MSEL scales.
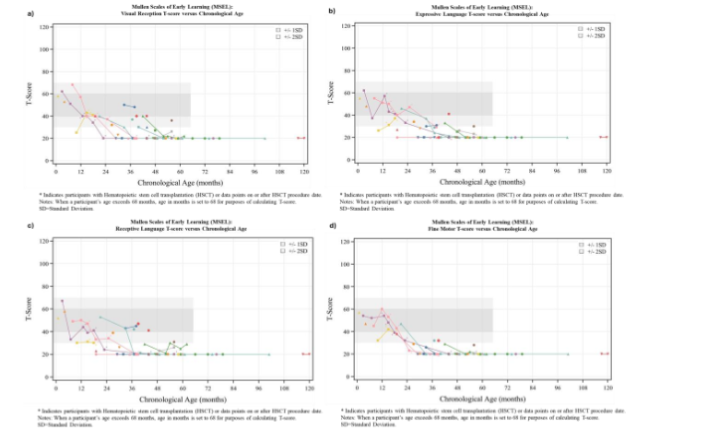
3.3 AGE WHERE DEVELOPMENT DEVIATES OUTSIDE NORMATIVE MEANS
Developmental trajectories deviated below -1SD from the normative mean at a mean chronological age range from 12.8 to 19.5 months. The earliest deviation from typical development by -1SD occurred with the Expressive Language scale at a mean chronological age of 12.8 months, followed by the Fine Motor scale (15.9 months), Receptive Language (17.4 months) and Visual Reception (19.5 months).
| Parameter | Visual Reception | Expressive Language | Receptive Language | Fine Motor |
|---|---|---|---|---|
| Chronological age (months) at inflection point | 70.9 | 67.0 | 74.6 | 66.1 |
| Developmental AEq score (months) at inflection point | 29.9 | 25.7 | 26.0 | 27.7 |
| Developmental AEq score (months) at inflection point upper 95% CI | 33.8 | 29.0 | 30.6 | 30.1 |
| Chronological age (months) when the developmental AEq mean trajectory deviates from the normative mean (below Mean -1SD bound) | 19.5 | 12.8 | 17.4 | 15.9 |
| Developmental AEq score when the mean trajectory deviates from the normative mean (below Mean -1SD) | 16.4 | 10.1 | 14.1 | 13.6 |
| Chronological age (months) when the developmental AEq mean trajectory deviates from the normative mean (below Mean -2SD bound) | 29.3 | 25.3 | 24.9 | 24.0 |
| Developmental AEq score when the mean trajectory deviates from the normative mean (below Mean -2SD bound) | 21.1 | 16.4 | 17.0 | 17.8 |
| Gain in AEq from -2SD to inflection point (Months AEq/52 Weeks Age) | 1.6 | 1.9 | 0.1 | 2.5 |
Developmental trajectories deviated below –2SD from the normative mean at a chronological age range from 24.0 to 29.3 months. The earliest deviation from typical development by –2SD occurred with the Fine Motor scale at a mean chronological age of 24.0 months, followed by Receptive Language (24.9 months), Expressive Language (25.3 months), and Visual Reception scale (29.3 months).
The mean chronological age where skill acquisition plateaued (inflection point) ranged by scale from 66.1 (Fine Motor) to 74.6 (Receptive Language) months, while the developmental AEq Score where skill acquisition plateaued ranged from 25.7 (Expressive Language) to 29.9 (Visual Reception) months.
The maximum developmental AEq score at the peak of the upper boundary of the 95% CI was 33.8 months for Visual Reception and the lowest developmental AEq score was 29.0 months for Expressive Language. The largest mean increase in skills per year from the timepoint where development is -2SD from the normative mean to the timepoint the plateau is reached was 2.5 months for Fine Motor and the smallest gain was 0.1 months for Receptive Language.
3.4 ESTIMATED MEAN AGE EQUIVALENCE CHANGE PER YEAR
Mean change was estimated for each MSEL scale AEq score after 1 and 2 years based on age at first assessment. Slowing in the rate of skill acquisition was present very early in development compared to normative peers across all MSEL scales, with skill acquisition per year at all age ranges being less than 12 months.
Mean rate of skill acquisition per year decreased as chronological age increased. The mean change in Visual Reception AEq per 12-month period decreased from 7.2 months at 6 to <9 months to 0.2 months at 63 to <66 months, indicating a slowing in the rate of skill acquisition compared to typically developing peers. This decrease in the rate of skill acquisition over time is a pattern seen in all other MSEL subscales.
The rate of skill acquisition slowed dramatically at a very young age on the Receptive Language scale. The mean gain at a chronological age between 18 to <21 months was 4.4 months per year and at a chronological age of 24 to <27 months there was a mean increase per year of only 3.9 months. The rate of skill acquisition varied by baseline function. For example, mean Visual Reception skill acquisition when developmental function was -1SD (chronological age 19.5 months) from the normative mean was 5.8 months/year, at -2SD (chronological age 29.3 months) was 4.7 months/year and, from -2SD to peak skill acquisition, the mean rate was 1.6 months/year.
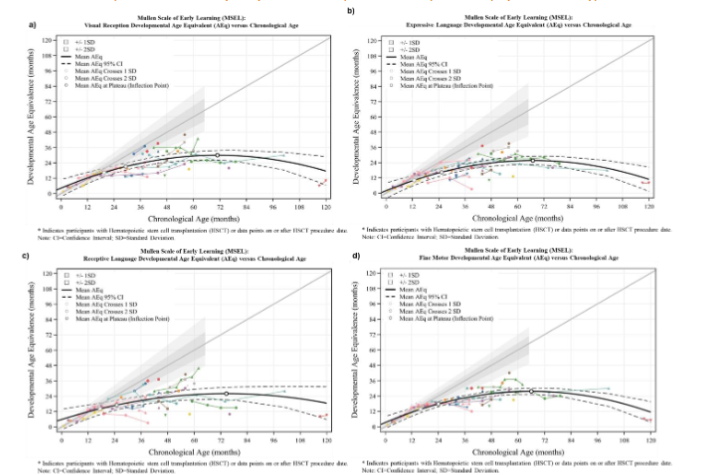
4. Discussion
We analyzed the developmental trajectory, rate of skill acquisition and timing of plateau in skill acquisition in a large group of patients using standardized measures. To better understand how the brain disease affects the development of pediatric patients with neuronopathic MPS II, it is important to evaluate function across multiple developmental domains. The neurodevelopmental data collected in this study were analyzed to understand developmental trajectories and inflection timepoints across the MSEL Visual Reception, Expressive Language, Receptive Language and Fine Motor scales. MSEL data in this study provide a comprehensive, multi-domain developmental picture of the differential neuronopathic involvement in MPS II, demonstrating that developmental delay is not uniform across scales. Several patients had developmental function on one MSEL scale above -1SD from the normative mean while other scales were below -2SD from the normative mean. This supports the importance of using a multi-domain measure to understand disease impacts and developmental variability in neuronopathic MPS II.
This study provides an important contribution to understanding the timing of the delay phase of development in neuronopathic MPS II with our results showing that a decline in MSEL T-scores is evident very early in development. Expressive Language and Fine Motor scales provided the earliest indication of delay and preceded a deviation from the normative mean for Visual Reception and Receptive Language. The mean gain per year in AEq for every MSEL scale at all age ranges illustrated that the rate of skill acquisition in children with neuronopathic MPS II slows early in life, skill acquisition per year is less than the change in chronological age, and the delay becomes more substantial over time. Mean skill acquisition did not peak until a chronological age of 66 to 75 months but the mean estimated rate of skill acquisition per year in Receptive Language was extremely reduced to 3.1 months at a chronological age of 33 months. These results support the importance of early treatment to reduce or prevent GAG accumulation at a time in development of exponential brain growth and to maximize function or halt developmental decline in a progressive neurodegenerative disease.
These data support that the rate of skill acquisition per year varies by baseline function and decreases further as chronological age increases. Where the child is in the disease continuum needs to be considered when interpreting a treatment response. Heterogeneity in function at baseline within a clinical study of children with neuropathic MPS II necessitates an understanding of estimated change per year that considers baseline disease severity. For example, 25 months is the mean age where expressive language deviates outside of -2SD from the normative mean. If a 25-month-old child has expressive communication skills that are lower than the mean trajectory and representative of function at -3SD, baseline disease severity more than chronological age should be considered in determination of an anticipated efficacy response. The magnitude of the anticipated efficacy response will be less with more brain neurodegeneration that is progressive and irreversible.
Similar developmental trajectories have been reported by Seo et al. using the Kyoto Scale of Psychological Development. In their study, 4 of 13 patients had developmental function at first available assessment below -2SD and only 1 of 4 patients had a developmental change ≥3 months AEq. The developmental function for children on standard of care with ERT peaked in the 2–3-year-old functional range. The developmental AEqs at the peak of the upper boundary of the 95% CI for the mean trajectory were 29.0 months (Expressive Language), 30.1 months (Fine Motor), 30.6 months (Receptive Language), and 33.8 months (Visual Reception). Thus, although children may acquire a small number of skills until they are 5–6 years old, function does not typically exceed the 2–3-year-old level and the rate of change per year is thereafter very small.
Patients entering the study could have been evaluated using any of several widely recognized neurodevelopmental assessments, but the MSEL was found to be the assessment predominantly used in patients enrolled in the study for which longitudinal assessment data were available. Kim et al. compared the AEq values for concurrently administered MSEL and BSID-III and found no statistical differences between mean subtest scores from patients with MPS II, IIIA, and IIIB. Overlap is present between many developmental concepts on the BSID-III and MSEL, with examples of developmental constructs included on both measures comprising visual attention, relational play, attention, counting, matching and sorting, spatial memory, expressive and receptive language, gross motor mobility, and fine motor grasp and dexterity. The MSEL has good psychometric properties in MPS disorders, with a test-retest reliability for a 1- to 2-week interval ranging from 0.84 to 0.96 and inter-rater reliability ranging from 0.91 to 0.99. MSEL convergent and divergent validity is present between children with typical development and autism and in children with neurodevelopmental conditions. Although the MSEL normative data were published in 1995 and have not been updated, a Flynn effect (increase in test scores) was not present for the typically developing children in either of these samples. The children with typical development in the autism study (age range from 16-40 months) and the neurodevelopmental conditions study (age 2-4 years) had T-scores within the expected normative range (mean 40, SD10) for their age, with mean T-scores of 49 to 53.
MSEL has been used to assess development in another rare disease, FMR1 (fragile X messenger ribonucleoprotein 1). Wheeler et al. used a very similar approach to this MPS II retrospective natural history study, using linear mixed modeling to explore developmental trajectories and to create developmental profiles. Similar to the present study, the MSEL was able to identify early developmental challenges and non-symmetrical developmental delay across scales.
A limitation of this retrospective study is that participants have been evaluated at different intervals and frequencies and data are both cross-sectional and longitudinal. Although there may be inconsistencies in long-term assessment data available across treatment centers, the neurodevelopmental data analyzed in this study were collected from a single clinical site, where MSEL raters had taken part in standardized administrative and scoring training for the MSEL within a translational research program. All data were reviewed, and quality was checked against MSEL manual values for T-scores and AEq scores. As with every study involving patients with rare diseases, recruitment was limited by the small sample size of the population; however, many patients were recruited from out of state representing a diverse demographic population. Longitudinal data collection is necessary to differentiate treatment response from natural history and although it would be optimal to develop a concurrent prospective study for comparisons of natural history and treatment response, it is ethically challenging to design comparative studies which would allow for the inclusion of non-treated control groups in fatal neurodegenerative rare conditions. This natural history retrospective data based on neurodevelopment assessments is therefore very valuable to understand the evolution of the disease.
5. Conclusion
Results of this study support that children with neuropathic MPS II present with developmental delay across multiple domains very early in development. Mean developmental trajectories support a deviation below -1SD as early as 12.8 to 19.5 months and below -2SD as early as 24.0 to 29.3 months. A multidomain assessment is beneficial to create a comprehensive neurodevelopmental picture and identify areas affected early as markers of initiation of neuronopathic MPS II. Rate of skill acquisition in the natural history of neuronopathic MPS II slows dramatically prior to a chronological age of 24 months and varies by where the patient is in the disease continuum. When designing endpoint models and responder definitions within a clinical study for MPS II, it is essential to understand the varied disease trajectories, inflection timepoints where function deviates outside of normal ranges, and how rate of skill acquisition varies by chronological ages and baseline function at the time of treatment.
Conflict of Interest Statement:
None
Funding
ME recruited the patients within the Program for Neurodevelopment in Rare Diseases at University of Pittsburgh Medical Center (UPMC). Funding for the data collection was secured from various institutions and multiple research activities at UPMC. The retrospective study was funded by REGENXBIO Inc. DP, YC, CM, MF are employed by REGENXBIO Inc.
The authors would like to thank the patients, families, healthcare professionals, and patient organizations who took part in this study. We would also like to thank Rare Disease Research Partners (RDRP) for medical writing support.
References
- D’Avanzo F, Rigon L, Zanetti A, Tomanin R. Mucopolysaccharidosis Type II: one hundred years of research, diagnosis, and treatment. Int J Mol Sci. Feb 13 2020;21(4)doi:10.3390/ijms21041258
- Wraith JE, Scarpa M, Beck M, et al. Mucopolysaccharidosis type II (Hunter syndrome): a clinical review and recommendations for treatment in the era of enzyme replacement therapy. Eur J Pediatr. 2008/03/01 2008;167(3):267-277. doi:10.1007/s00431-007-0635-4
- Young ID, Harper PS, Newcombe RG, Archer IM. A clinical and genetic study of Hunter’s syndrome. 2. Differences between the mild and severe forms. J Med Genet. Dec 1982;19(6):408-11. doi:10.1136/jmg.19.6.408
- Holt J, Poe M, Escolar M. Natural progression of neurological disease in mucopolysaccharidosis type II. Pediatrics. 05/01 2011;127:e1258-65. doi:10.1542/peds.2010-1274
- Shapiro EG, Jones SA, Escolar ML. Developmental and behavioral aspects of mucopolysaccharidoses with brain manifestations – neurological signs and symptoms. Mol Genet Metab. Dec 2017;122S:1-7. doi:10.1016/j.ymgme.2017.08.009
- Holt J, Poe MD, Escolar ML. Early clinical markers of central nervous system involvement in mucopolysaccharidosis type II. J Pediatr. 2011/08/01/ 2011;159(2):320-326.e2. doi:https://doi.org/10.1016/j.jpeds.2011.03.019
- Link B, de Camargo Pinto LL, Giugliani R, et al. Orthopedic manifestations in patients with mucopolysaccharidosis type II (Hunter syndrome) enrolled in the Hunter Outcome Survey. Orthop Rev (Pavia). Sep 23 2010;2(2):e16. doi:10.4081/or.2010.e16
- Shapiro EG, Eisengart JB. The natural history of neurocognition in MPS disorders: a review. Mol Genet Metab. May 2021;133(1):8-34. doi:10.1016/j.ymgme.2021.03.002
- Phillips D, Cho Y, Mulatya C, Nevoret M-L, Poe M, Escolar M. Natural history of neurodevelopment in neuronopathic mucopolysaccharidosis type II (MPS II): Mullen Scales of Early Learning (MSEL) cognitive, motor and language developmental trajectories. Mol Genet Metab. 02/01 2022;135:S97. doi:10.1016/j.ymgme.2021.11.253
- Barone R, Pellico A, Pittala A, Gasperini S. Neurobehavioral phenotypes of neuronopathic mucopolysaccharidoses. Ital J Pediatr. Nov 16 2018;44(Suppl 2):121. doi:10.1186/s13052-018-0561-2
- Jones SA, Almássy Z, Beck M, et al. Mortality and cause of death in mucopolysaccharidosis type II – a historical review based on data from the Hunter Outcome Survey (HOS). J Inherit Metab Dis. Aug 2009;32(4):534-43. doi:10.1007/s10545-009-1119-7
- Parini R, Rigoldi M, Tedesco L, et al. Enzymatic replacement therapy for Hunter disease: up to 9 years experience with 17 patients. Mol Genet Metab Rep. Jun 2015;3:65-74. doi:10.1016/j.ymgmr.2015.03.011
- Scarpa M, Almássy Z, Beck M, et al. Mucopolysaccharidosis type II: European recommendations for the diagnosis and multidisciplinary management of a rare disease. Orphanet J Rare Dis. 2011/11/07 2011;6(1):72. doi:10.1186/1750-1172-6-72
- Janzen D, Delaney KA, Shapiro EG. Cognitive and adaptive measurement endpoints for clinical trials in mucopolysaccharidoses types I, II, and III: a review of the literature. Mol Genet Metab. Jun 2017;121(2):57-69. doi:10.1016/j.ymgme.2017.05.005
- Mullen EM, American Guidance S. Mullen Scales of Early Learning. AGS ed ed. AGS Circle Pines, Minnesota; 1995.
- Wheeler AC, Gwaltney A, Raspa M, et al. Emergence of developmental delay in infants and toddlers with an FMR1 mutation. Pediatrics. May 2021;147(5)doi:10.1542/peds.2020-011528
- Cho Y, al. e. Beyond the normative data: understanding the Mullen Scales of Early Learning. ICIEM Virtual Congress. 2021.
- Seo JH, Okuyama T, Shapiro E, Fukuhara Y, Kosuga M. Natural history of cognitive development in neuronopathic mucopolysaccharidosis type II (Hunter syndrome): contribution of genotype to cognitive developmental course. Mol Genet Metab Rep. Sep 2020;24:100630. doi:10.1016/j.ymgmr.2020.100630
- Kim A, Poe M, Escolar M. Mullen Scales of Early Learning (MSEL) and Bayley Scales of Infant and Toddler Development (BSID): utility in assessing cognitive endpoints in MPS clinical trials. Mol Genet Metab. 2021;132(2):S55.
- van der Lee JH, Morton J, Adams HR, et al. Cognitive endpoints for therapy development for neuronopathic mucopolysaccharidoses: Results of a consensus procedure. Mol Genet Metab. Jun 2017;121(2):70-79. doi:10.1016/j.ymgme.2017.05.004
- Akshoomoff N. Use of the Mullen Scales of Early Learning for the assessment of young children with Autism Spectrum Disorders. Child Neuropsychol. Aug 2006;12(4-5):269-77. doi:10.1080/09297040500473714
- Burns TG, King TZ, Spencer KS. Mullen scales of early learning: the utility in assessing children diagnosed with autism spectrum disorders, cerebral palsy, and epilepsy. Appl Neuropsychol Child. 2013;2(1):33-42. doi:10.1080/21622965.2012.682852