






Pathology of Celiac Disease: Diagnosis and Mechanisms
Pathology of celiac disease: An insight about etiopathogenesis, pathophysiology and histologic diagnosis
Introduction
Celiac disease (CD) is an immune mediated disorder characterized by intolerance to glutens in certain grains like wheat, barley, and rye. The gliadin exposure in susceptible individuals leads to an inflammatory reaction with damage to the small intestinal mucosa with villous blunting, atrophy and ultimate disappearance of intestinal villi. The damaged intestinal villi lead to malabsorption with diarrhea, steatorrhea, weight loss, fatigue, and abdominal pain. The CD is unique amongst autoimmune disorders. The typical autoimmune features include a strong human leucocyte antigen (HLA) association, production of specific antibodies, and immune-mediated killing of a particular cell type (intestinal cells). The uniqueness lies in the fact that the disease only manifests upon exposure to an exogenous factor.
The duodenal biopsy for histologic assessment remains the gold standard in the diagnosis of CD. The histologic changes in the duodenum include villous atrophy associated with increased intraepithelial lymphocytes. As similar changes can be seen in various other pathological conditions, the accurate diagnosis of CD involves integration of clinical, serological and genetic characteristics in the histologic assessment of CD.
The clinical presentation of celiac disease can be variable. In the mild form, patients can be almost asymptomatic whereas in the most severe form of disease patients may present with many complications which may be life threatening¹⁻³.
The present article describes in detail etiopathogenesis, pathophysiology, and pathologic features of CD. Better understanding of these features is not only important for correct diagnosis but also results in better patient management and would pave a way for future research of this overly complex disease characterized by complex interaction of environmental, genetic as well as immunologic mechanisms.
Pathogenesis
The development of celiac disease (CD) is determined by both environmental and genetic factors. The gluten in the grains represents an environmental factor. In addition to this environmental factor, CD development involves genetic predisposition as most CD patients possess human leucocyte antigen (HLA) DQ2 and/or HLA-DQ8. In CD, CD4 + T-cell response is raised against several distinct gluten peptides and these peptides are recognized in the context of CD-associated HLA DQ molecules. In addition, the patients make antibodies specific for gluten proteins. The complex interplay of multiple genetic and environmental factors determines the development of CD.
ENVIRONMENTAL FACTORS:
Gluten
Gluten is the name applied to a set of proteins found in grains of wheat, barley, and rye⁴⁻⁵. Gluten proteins are rich in glutamine and proline residues. In wheat, gluten proteins are divided into gliadins and glutenins. Each wheat variety expresses multiple α β and ω gliadins in addition to low and high molecular weight glutenins. While gliadins are soluble in alcohol, glutenins are not soluble in alcohol. Gluten has an extremely high content of amino acids glutamine (30%) and proline (15%). High proline content renders gluten highly resistant to degradation by gastrointestinal enzymes, making it possible for large immunogenic gluten peptides to reach the mucosal surface⁶⁻⁷. There may be up to hundred different gluten peptides in a single wheat variety and many of these are implicated in the pathogenesis of celiac disease. Gluten proteins of barley, and rye are termed hordeins and secalins, respectively. Gut lesions of most patients with CD completely resolve when gluten is excluded from the diet, and they reappear when the patients again consume gluten.
Non-gluten factors
Environmental factors other than glutens have been suspected in the development of CD as only minority of individuals who carry HLA DQ2 and HLA DQ8 haplotypes ever develop the disease. Infections early in life are suspected and there appears to be role of Rota virus infection based on epidemiological evidence. Some studies suggested that rotavirus vaccination may protect against development of CD⁸⁻⁹. There is some evidence for association of CD with other infectious agents including Helicobacter pylori, adenovirus, and enterovirus¹⁰.
GENETIC FACTORS:
HLA – genes
CD is a highly heritable disease as indicated by strong familial clustering and a high concordance rate in monozygotic twins¹¹˒¹². The genes leading to susceptibility for CD have been mapped with Genome-Wide association studies (GWAS). There is strong genetic influence in CD as the concordance between monozygotic twin is 80% but only 11% in dizygotic twins⁸ which is the same as the risk for first degree relative¹³. There is a strong association between CD and the HLA complex. The strongest association is observed with HLA DQ 2.5; more than 90% celiac patients possess one or two copies of HLA DQ 2.5¹⁴. CD is associated to a lesser extent with HLA DQ8¹⁵. Individuals homozygous for HLA DQ 2.5 have a fivefold increased risk for development of CD compared to individuals heterozygous for HLA DQ 2.5¹⁶. Another HLA – DQ variant – HLA DQ 2.2 has a peptide binding motif almost identical to that of HLA DQ 2.5¹⁷. Whereas HLA – DQ 2.5 predisposes to CD, HLA – DQ 2.2 does not. This difference is related to the peptide – binding properties of these HLA – DQ variants. The estimated risk effect of HLA – DQ2 and HLA DQ8 on CD development is estimated to be about 35%¹⁸. Not all HLA and DQ 2.5+ and HLA DQ 8+ individuals develop CD indicating that these HLA genotypes are necessary but not sufficient for the development of CD. Recently many additional genetic loci for CD have been identified. Most of these genes encode proteins involved in immunity in supporting the notion that CD is an immune mediated disorder.
Gut Immune System and adaptive immune response in celiac disease
GUT IMMUNE SYSTEM
The gut immune system involves both innate and adaptive parts and can be divided into geographically distinct inductive and effector sites. The inductive sites comprise gut-associated lymphoid tissue (GALT), specifically Peyer’s patches and isolated lymphoid follicles. In addition, antigens taken up from the lumen can initiate immune response in gut – draining lymph nodes¹⁹. In these structures, priming of B and T cells, as well as memory recall responses take place. In the GALT, B cells diversify their BCRs through somatic hypermutation and differentiate into long-lived plasma cells and memory B cells. The gut plasma cells display more mutation in their BCRs than any other B cells or plasma cell population in the body²⁰. After antigen stimulation, the prime cells travel via thoracic duct and reach blood circulation and then via blood populate the epithelium (T cells) and lamina propria (T cells and plasma cells) as effector cells protecting the tissue against microbial intruders. These effectors are tissue resident and extremely long lived²¹⁻²³.
INTRAEPITHELIAL LYMPHOCYTES
The intraepithelial lymphocyte (IEL) compartment is composed of T cells expressing either αβ or nΔ T -cell receptors with either helper (CD 4⁺) or suppressor (CD 8⁺) phenotypes. In celiac disease, the number of IEL is increased from a normal number of < 25 IELs per hundred enterocytes and although not unique to CD, is considered a diagnostic sign of the disease. Many of these T cells also express natural killer (NK) cell receptor – both activating and inhibitory receptors. NK cell receptors are believed to be responsible for killing of enterocytes which is an essential component of celiac disease²⁴⁻²⁶.
LAMINA PROPRIA T CELLS
In active CD, there is a marked infiltration of TCR αβ⁺ T cells in the lamina propria. Unlike the IELs both CD 4⁺ and the CD 8⁺ T cells in the lamina propria lack the proliferation markers Ki67 in active CD²⁷. In active CD, fraction of CD4⁺ T cells express markers associated with a subset of regulatory T cells; however, it is not clear whether regulatory CD 4⁺ T cells play a role in the pathogenesis of CD²⁸.
LAMINA PROPRIA PLASMA CELLS
The active CD is associated with lamina propria plasmacytosis with an increased density of immunoglobulin A (IgA), IgM, and IgG plasma cells by factors of 2.4, 4.6, and 6.5²⁹. In normal small intestine, IgA expressing plasma cells dominate with very few IgG cells. In contrast to IgG plasma cells, IgA and IgM plasma cells express surface immunoglobulins³⁰˒³¹. These molecules act as functional B cell receptors (BCR). In addition to BCR, gut plasma cells express HLA class II molecules, raising the possibility that they might have role as antigen presenting cells³².
LAMINA PROPRIA MYELOID CELLS
The CD lesion has increased density of neutrophils and monocyte – derived cells. In active CD, infiltration of neutrophils in the lesion has been demonstrated³³˒³⁴.
LAMINA PROPRIA STROMAL CELLS
Lamina propria stromal cells such as fibroblasts and myofibroblasts are important for building the extracellular matrix. In addition, they are also involved in creating niches for the intestinal stem cells, maturation of the epithelial cells along the crypt-villous axis and have various immune functions like bone marrow where they are important for creating survival niches for long lived plasma cells³⁵. Stromal cells in the lamina propria may support long-lived plasma cells in the gut³⁶. In relation to CD, knowledge how the composition and function of lamina propria stromal cells may be altered is limited.
LOCAL CYTOKINE/CHEMOKINE ENVIRONMENT
The understanding of local cytokine and chemokines in the context of CD is limited. Gluten specific CD4⁺ T cells produce IFN – γ³⁷ and intraepithelial TCR αβ⁺ CD 8 and TCR delta γ T cells also likely contribute with production of this cytokine²⁴. The chemokine CXCL 10 and CXCL 11 are produced by enterocytes in response to IFN – γ stimulation³⁸ and they play a role in migration of lymphocytes in the epithelium. Increased expression of IL-15 is observed in CD lesion in both the epithelium and lamina propria³⁹. The cytokine stimulates proliferation and survival of T-cells and NK cells⁴⁰ and promotes tissue destruction⁴¹.
ADAPTIVE IMMUNE RESPONSE
CD develops almost exclusively in HLA – DQ 2.5 or HLA – DQ 8 positive individuals. Gluten – derived peptides presented by HLA DQ 2.5 or HLA DQ 8 induce CD4⁺ T cells response. HLA – DQ 2.5 and HLA – DQ 8 prefer to bind peptides with negatively charged aminoacids at anchor residues; however, gluten peptides do not carry negative charge and therefore bind poorly to HLA – DQ 2.5 or HLA DQ 8. Tissue transglutaminase 2 (TG2) can convert non-charged glutamine in gluten into negatively charged glutamic acid (deamidation). This increases binding capacity of gluten peptides to HLA – DQ 2.5 and DQ 8⁴²˒⁴³. TG2 for most parts remains intracellularly in an inactive form and is activated upon its release during tissue damage⁴⁴˒⁴⁵. Tjon et al proposed two possible mechanisms which could trigger tissue damage. CD4⁺ T cell response against native gluten peptide could represent the first breach in oral tolerance to gluten. The presentation of native gluten peptides by HLA – DQ 2 and HLA – DQ DQ 8 to CD 4⁺ T-cells leads to production of IFN – γ. IFN – γ in turn leads to higher expression of HLA – DQ molecules thereby increasing gluten peptide presentation. In presence of gluten this could become a self amplifying loop causing some tissue damage. The tissue damage would lead to release of TG2which would modify gluten peptides into high affinity ligands for HLA – DQ 2 and/or HLA – DQ8, by process of deamidation. The gluten specific CD4⁺ T – cell response is thus expanded leading to additional tissue damage with initiation of second self amplifying loop. The second possible mechanism for tissue damage might be gastrointestinal tract infections⁴⁶.
DAMAGE TO INTESTINAL EPITHELIUM
Intraepithelial lymphocytes (IEL) are localized between intestinal epithelial cells and play an important role in immunosurveillance of the epithelium. IEL represent a mixed population of TCR αβ⁺ T cells, TCR γΔ⁺ T cells and NK cells with vast majority of IELs being CD 8⁺ TCR αβ⁺ T cells⁴⁷. The NK – cell receptors expressed on TCR IELs are distinct from NK – cell receptors expressed on blood T cells⁴⁷. The NK cell receptors are thought to act mainly as T cell costimulators lowering the threshold for T-cell activation in stressful times⁴⁸.
In active CD, CD8⁺ TCR αβ⁺ and TCR γΔ⁺ IELs are markedly increased. Akin to normal controls, IELs from active celiac disease express NK cell receptors. However, as compared to normal controls, IELs from active CD acquire more activating NK-cell receptors⁴⁷. In healthy small intestine, IELs predominantly express inhibitory receptors. Interaction of activating NK-cell receptors with their ligands will enhance IFN-γ production and cytolysis leading to tissue damage. An important factor in acquiring an activating NK cell receptor repertoire is interleukin 15 which has been shown to upregulate both NKG2D and CD94/NKG2C on IELs of active CD patients and boost their ability to lyse enterocytes²⁵. IELs can also alter the NK-cell receptor function leading to NK-cell receptor mediated cytotoxic independent of TCR specificity. While gluten-specific CD4⁺ T cells elicit an inflammatory response in the lamina propria, IELs in the epithelium acquire activating NK receptors and the ability to lyse stressed epithelial cells independent of T-cell receptor signalling which likely contributed to the tissue damage in CD.
In refractory celiac disease, the survival, expansion, and acquisition of NK cell like phenotype is even more pronounced than in CD, probably due to presence of larger amounts of IL-15. RCDI patients have an aberrant clonal IEL population that lacks surface TCR-CD3 expression. Studies on aberrant TCR-CD3 negative IEL lines from RCD II patients showed that upon stimulation with IL-15, these cell lines express granzyme β and lyse intestinal epithelial cell line HT29. This study suggests a role of abnormal IEL in perpetuating epithelial damage in RCD II patients³⁹. IL-15 dependent NK cell like transformation of IELs may be an essential step in the immunopathology of RCD.
Possible sequence of events in the development of celiac disease
Tjon et al suggested possible sequence of events in the development of celiac disease⁴⁶(Table 1). The healthy individuals may have antibodies against gluten peptide and CD4⁺ T-cells specific for native gluten peptide; however, regulatory processes in the intestine keep gluten specific T cell response in check. This steady state can be breached by frequent episodes of enteroviral infections leading to secretion of inflammatory cytokines and differentiation of Th 1 cells with enhanced response to gluten⁴⁹. The low level of gluten reactivity and pathogen induced inflammation together could lead to tissue damage and release of TG2. The TG2 would generate a large repertoire of deamidated gluten peptides with high affinity for HLA DQ with boosting of gluten-specific T cell response. Regulatory processed are no longer able to contain T cell response and exposure to gluten leads to inflammation. At this point return to the steady state is possible only with the elimination of gluten. HLA DQ phenotype always plays an important role in the development of CD⁴⁶.
Table 1: Possible sequence of events in the development of celiac disease
| Healthy individuals: | May have antibodies against gluten peptide and CD4⁺ T-cells specific for native gluten peptides. Regulatory processes in intestine keep gluten specific T cell response in check. |
|---|---|
| Breaching of steady- state: | Frequent episodes of enteroviral infections → secretion of inflammatory cytokines and differentiation of Th 1 cells with enhanced response to gluten → Tissue damage and release of TG2 → Large repertoire of deamidated gluten peptides with high affinity for HLA DQ → Boosting of gluten specific T cell response. Regulatory processed are no longer able to contain T cell response and exposure to gluten leads to inflammation. |
| Return to the steady state: | Possible only with the elimination of gluten. |
Note: At all stages, HLA DQ phenotype plays an important role in the development of celiac disease.
Pathology
ENDOSCOPIC APPEARANCE
The celiac disease may be associated with mucosal abnormalities suspicious of CD. This should lead to duodenal biopsies even in the absence of classic symptoms of CD. The endoscopic abnormalities include decreased or absent Kerckring’s folds⁵⁰, mosaic pattern, scalloped folds, and visible underlying blood vessels⁵¹. The endoscopic findings of ulceration, stricture usually indicate complicated CD, inflammatory bowel disease, or malignancy. The presence of normal duodenal mucosa does not rule out CD⁵².
BIOPSY RECOMMENDATIONS
Small bowel mucosal biopsies remain gold standard for the diagnosis of CD because of variation in the clinical findings and serologic testing⁵²˒⁵⁹. The optimum number of biopsies needed for the diagnosis of CD is not fully established. The pathological findings in CD can be patchy and can affect different areas of duodenum with varying degrees of severity. Therefore, multiple biopsies of the duodenum (at least four) should be performed if CD is suspected on clinical grounds. The American Gastroenterology Association recommends 6 biopsies from the distal duodenum for the diagnosis of CD⁵⁵ or 4 biopsies from the distal duodenum with 2 biopsies from the duodenal bulb as an alternative approach⁶⁰. The strict adherence to these guidelines in clinical practice is suboptimal (about 35%), although strict adherence to these guidelines increases the detection of celiac disease⁶¹˒⁶². An accurate diagnosis of celiac disease in suspected cases has 2 important prerequisites ; biopsies should be performed when the patient consumes gluten containing diet and biopsy tissue fragments are well-oriented.
NORMAL HISTOLOGY OF DUODENUM AND JEJUNUM
In a well-oriented biopsy cut at right angle well formed, long, slender villi are seen projecting from a simple gland layer into the lumen. The glands are orderly arranged, regularly spaced, and rest on the muscularis mucosae (Figure 1). The normal crypt to villous ratio is about 1:3-4 in adults and about 1:2 in children. The villi are lined by columnar epithelium with brush border which can be appreciated in the hematoxylin and eosin (Ha and E) stained sections as a faint density. The villous epithelium is continuous with the crypt epithelium which represents the regenerative zone. The villous epithelium is composed of absorptive cells intermixed with variable number of goblet cells. Paneth cells and occasional enterochromaffin cells are scattered in the crypts. Mitoses are seen in the crypt epithelium but are normally not seen in the villous epithelium.
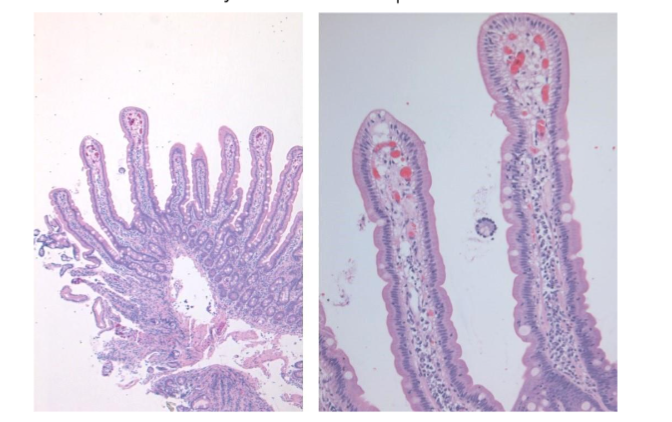
Figure 1: Normal duodenal mucosa showing long, slender, regularly spaced villi with orderly arrangement. The normal crypto to villous ratio of 1:3 – 4 is seen. The villi are lined by columnar epithelium with brush border which can be seen as a faint density on the luminal aspect.
Occasional IELs can be seen in normal small bowel epithelium which are part of gut-associated lymphoid system including gut associated lymphoid follicles, Peyer’s patches, and mesenteric lymph nodes. In normal small bowel mucosa, IELs demonstrate a “decrescendo” pattern. i.e. the density of IELs in the tips of the villi is lower than that in the proximal third of villi⁶³. In the jejunum, there are about 6 to 40 lymphocytes per 100 villous epithelial cells in adults⁶⁴ and 12 to 35 per 100 in children⁶⁵. In the duodenum IEL count is lower than those in jejunum. The upper limit of normal IEL count in the duodenum is 20 to 25 IELs per 100 epithelial cells.
The lamina propria of the villous core and surrounding mucosa shows small number of lymphocytes with occasional plasma cells, eosinophils, mast cells and histiocytes. Brunner’s glands are seen in the duodenum but variably in the jejunum. These can be seen on either side of the muscularis mucosae. Prominence or hyperplasia of Brunner’s glands is usually associated with broad and short villous architecture.
MICROSCOPIC FEATURES OF CELIAC DISEASE
The key microscopic features of celiac disease include varying degree of villous atrophy, crypt hyperplasia, chronic inflammatory infiltrate, and increased IELs. The overall mucosal thickness is normal or only slightly decreased. The degree of villous blunting can be variable ranging from minimal blunting to completely flat mucosa⁵³˒⁵⁴. Accurate assessment of villous blunting needs proper orientation of the biopsies. The biopsies however are seldom well oriented; in such cases, attention should be made to find 3 to 4 well oriented villi for architectural assessment. The lamina propria contains increased inflammatory infiltrate consisting of lymphocytes, plasma cells, with some eosinophils, histiocytes, and basophils.
Various classification schemes have been developed for assessing the severity of CD. The original Marsh classification scheme recognises 4 types ; an infiltrative lesion (Marsh Type 1), hyperplastic lesion (Marsh Type 2), and destructive lesion ( Marsh Type 3). The original Marsh classification does not mention cut off value for the number of IELs. The updated Marsh classification proposed 40 IELs /100 enterocytes as the upper cutoff point for normal and further subdivided group 3 into 3a, 3b, and 3c based on severity of villous shortening or blunting⁶⁶. (Table 2). Marsh has strongly argued against the technical basis for this division⁶⁷. The updated Marsh classification known as the Marsh – Oberhuber classification system describes 5 histologic lesions; preinfiltrative (type 0), infiltrative (type 1), infiltrative – hyperplastic (type 2), flat – destructive (type 3), and atrophic – hypoplastic (type 4).
Table 2. Marsh-Oberhuber classification of Celiac Disease by Oberhuber et al
| Type | Intraepithelial lymphocytes per hundred enterocytes | Crypt | Villi |
|---|---|---|---|
| Preinfiltrative (Type 0) | Normal (< 40) | Normal | Normal |
| Infiltrative (Type 1) | > 40 | Normal | Normal |
| Infiltrative hyperplastic (Type 2) | > 40 | Hyperplastic | Normal |
| Destructive type 3a | > 40 | Hyperplastic | Mild blunting |
| Destructive type 3b | > 40 | Hyperplastic | Moderate blunting |
| Destructive type 3c | > 40 | Hyperplastic | Severe blunting (Flat) |
| Hypoplastic type 4 | > 40 | Atrophic | Severe blunting (Flat) |
Preinfiltrative lesions (type 0) are associated with normal mucosa seen mainly in patients with dermatitis herpetiformis with no evidence of malabsorption. Infiltrative lesions (type 1) show an increased IELs with preserved villous architecture (Figure 2) while infiltrative – hyperplastic lesions (type 2) show in addition crypt hyperplasia. Both patterns can be seen in patients with CD without symptoms, patients with dermatitis herpetiformis, and in first degree relatives of patients with CD. Destructive lesions (type 3), show villous blunting accompanied by crypt hyperplasia and IEL with variable degree increased chronic inflammation in the lamina propria (Figure 3). The mucosal thickness is normal or near normal. Hypoplastic lesions (type 4), refer to an atrophic small bowel mucosa with complete villous blunting and crypt atrophy (Figure 4, 5).
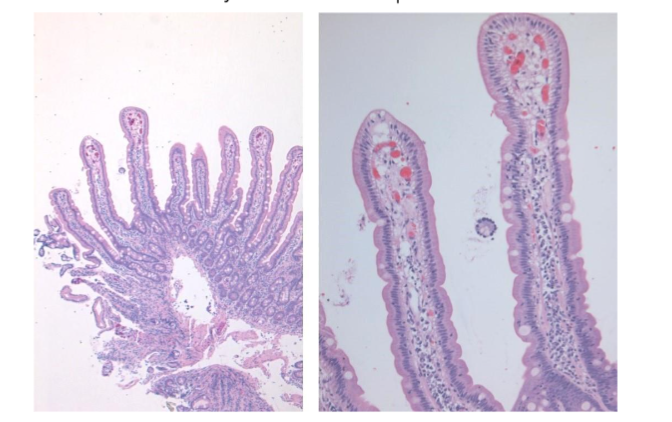
Figure 2: Infiltrative lesion. Normal villous architecture with increased intraepithelial lymphocytes. CD 3 immunostain highlighting the intraepithelial T lymphocytes.
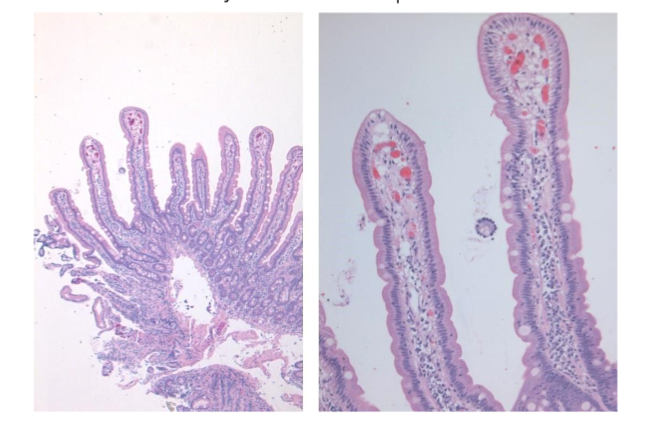
Figure 3: Subtotal villous atrophy. Atrophic small bowel mucosa with villous blunting and increased mucosal chronic inflammation with increased intraepithelial lymphocytes.
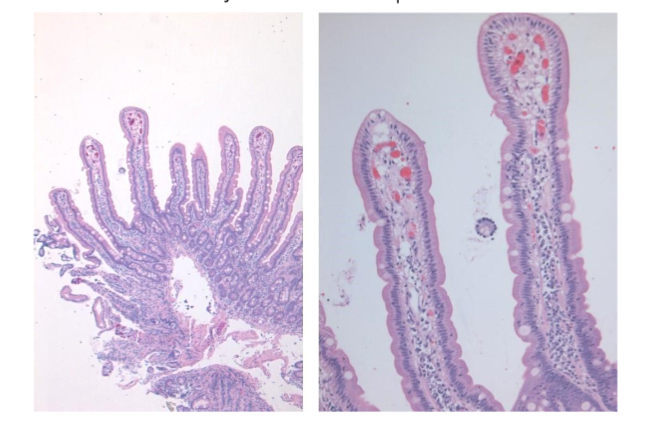
Figure 4: Celiac disease with complication. The patient presenting with intestinal obstruction. Resected segment of small intestine showing complete mucosal atrophy with flattened mucosa with loss of mucosal pattern.
Figure 5: Hypoplastic lesion with atrophic small bowel mucosa with complete loss of villous architecture and flattened mucosa and crypt atrophy.
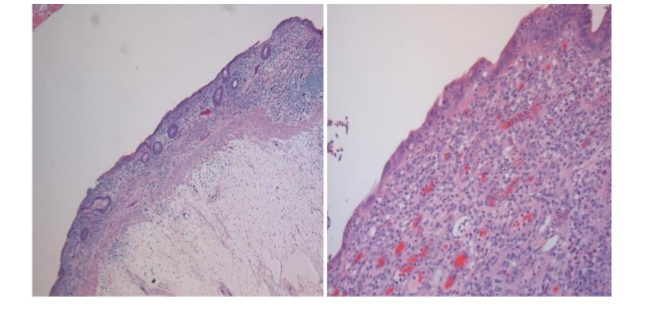
Corazza – Villanacci scoring system divided CD into 3 grades: grade A, grade B1, and grade B2⁶⁸ (Table 3). Grade A shows normal crypts and villous architecture but increased IELs (> 25 IELs/100 enterocytes). Grade B1 shows atrophic but detectable villi and increased IELs, and grade B2 shows flat/undetectable villi and increased IELs. This scoring system has less variability and greater agreement between pathologists⁶⁸.
Table 3. Corazza-Villanacci Score of Celiac Disease (Corazza et al)
| Grade | Intraepithelial lymphocytes per 100 enterocytes | Villi |
|---|---|---|
| A | > 25 | Normal |
| B1 | > 25 | Atrophic but detectable |
| B2 | > 25 | Flat/undetectable |
ASSESSMENT OF RESPONSE TO GLUTEN FREE DIET (GFD)
The standard treatment of celiac disease with demonstrated villous atrophy is the institution of the GFD. This should result in rapid alleviation of symptoms and return of abnormal serology to normal range. The histological evidence of response with restoration of villous architecture and reduction of mucosal inflammation is important as negative celiac serology does not guarantee mucosal healing⁶⁹. In a recent meta-analysis, the sensitivity of tTG for detection of persistent villous atrophy was only 50%.
Some patients fail to respond to GFD often because gluten is not adequately excluded from the diet. The second reason may be very severe initial lesion with completely flat mucosa. As IELs remain elevated for many years even in patients with GFD, assessment of the response to GFD relies on villous architecture. Unfortunately, studies detailing histological follow-up of celiac patients on GFD are inconsistent with respect to the response⁷⁰. One factor contributing to this variance could be inconsistency in the reporting of the response to a GFD. To address this issue, quantitative approaches such as Q-MARSH have been proposed. “Normalization” is defined as VH/Cd ratio of >2.5:1⁷¹.
It is likely that 1 year is too short a follow-up for histology, and that a longer period of follow-up is required before labelling the patient as having refractory celiac disease. In one series, persistent villous blunting was present at 1 year; this reduced to 5% over 2 – 5 years of follow-up⁷².
Histopathological differential diagnoses of celiac disease
Depending upon the histologic pattern seen in CD i.e. increased IELs or villous blunting differential diagnoses may vary; nevertheless, it is important to rule out conditions with similar morphologic changes in the small intestine (Table 4).
Table 4. Differential Diagnoses of Celiac Disease
Immunodeficiency
- Immunodeficiency
- Common variable immunodeficiency
- Infantile X-linked immunodeficiency
- Selected IgA deficiency
- T-cell deficiency or severe combined immunodeficiency
Infection
- Helicobacter pylori gastritis
- Tropical sprue
- Bacterial overgrowth
- Viral gastroenteritis or postinfective malabsorption syndrome
- Parasitic infestation
Diet, nutrition, and medication
- Food allergy
- Nutritional deficiency
- Morbid obesity
- Medication (including nonsteroidal anti-inflammatory drugs) and peptic injury
Autoimmunity
- Refractory sprue
- Autoimmune enteropathy
- Crohn disease, ulcerative colitis
- Lymphocytic or collagenous colitis
- Autoimmune diseases
Immunodeficiency
INFANTILE X-LINKED IMMUNODEFICIENCY:
Small bowel biopsy usually shows sparsity or absence of mucosal plasma cells. The mucosa associated lymphoid tissue shows lack of germinal centres⁷³˒⁷⁴. The small intestinal villous architecture is usually maintained.
COMMON VARIABLE IMMUNODEFICIENCY (CVID):
CVID is a heterogenous condition with many common features such as recurrent infections, autoimmunity and lymphoproliferation and laboratory evidence of hypergammaglobulinemia⁷⁰. The most common gastrointestinal symptom is diarrhea (92%), and evidence of malabsorption is seen in 54% patients of CVID in a large cohort study⁷⁵. Histological examination showed IELs in approximately 75.6 % cases, while villous atrophy was present in 51% cases⁷⁵. These cases however showed marked depletion of plasma cells and follicular lymphoid hyperplasia⁷⁵. Features which may be helpful in distinguishing CD from CVID associated enteropathy are profound depletion of plasma cells and follicular lymphoid hyperplasia as these 2 features are present in 96.3 % and 55 % cases of enteropathy associated CVID respectively. Celiac serology may also be helpful as only 11.5% of enteropathy associated CVID had celiac antibodies.
SELECTIVE IGA DEFICIENCY
The most common intestinal symptoms include diarrhea, and steatorrhea. The gastrointestinal symptoms may be due to parasitic infestation – giardiasis, CD, or milk intolerance. In some patients CD like mucosal lesions such as villous blunting and IELs are seen⁷⁶.
IgG DEFICIENCY
Chronic diarrhea, villous blunting and crypt hyperplasia may be seen in patients with IgG deficiency. Perlmutter et al in their study these features in 50% of infants⁷⁷.
T CELL DEFICIENCY OR SEVERE COMBINED IMMUNODEFICIENCY
The jejunal biopsy may features akin to CD with villous blunting, crypt hyperplasia, IELs, mucosal edema. The mucosa however may show many foamy macrophages with material stainable with PAS, toluidine blue and Giemsa stain⁷⁸.
Infection
HELICOBACTER PYLORI INFECTION
H. pylori infection may be associated with IELs but there is no or at the most only minimal villous blunting. The mucosa also shows considerable acute inflammation and there may be associated foveolar gastric metaplasia. The definite diagnosis requires assessment of gastric biopsies or demonstration of H. pylori by some other methods. Coexisting H. Pylori infection and CD have been reported.
TROPICAL SPRUE
There is a considerable overlap between clinical presentation and endoscopic appearance of tropical sprue and CD. The tropical sprue is usually associated with milder villous abnormalities and IELs tend to concentrate in the basal one third of the villi or the mucosal crypts⁷⁹. Tropical sprue tends to be patchy and shows more eosinophils⁷⁹. Other helpful features to differentiate from celiac include history of travel to the endemic areas, negative celiac serology, and good response to antibiotics and folic acid.
BLIND LOOP SYNDROME
In the blind loop syndrome bacteria colonise small intestine and with this mucosa may show changes akin to CD. The changes noted however are patchy in distribution and usually are of mild to moderate degree with slight to moderate villous atrophy. There may be history of intraabdominal surgery or bowel obstruction.
VIRAL OR BACTERIAL INFECTION
Several viral infections including human immunodeficiency virus are known to produce villous blunting and intraepithelial lymphocytosis. The mucosal changes are usually mild to moderate; however very severe changes with marked villous abnormalities resembling CD can occasionally be seen. The inflammatory infiltrate is predominantly acute with numerous neutrophils associated with crypt abscesses. There may be history of acute onset gastroenteritis and mucosal abnormalities return to normal with resolution of symptoms. There is direct evidence that celiac-like enteropathy can be initiated by norovirus infections and other viral infections⁸⁰˒⁸¹.
PARASITIC DISEASE
Malabsorption associated with mucosal abnormalities with villous atrophy and crypt hyperplasia may be seen in parasitic diseases. The lamina propria inflammatory infiltrate is usually rich in eosinophils in parasitic infestations. The villous abnormality is seen at a very low rate. This means if villous abnormality comparable to that seen in CD is noted in association with parasitic organisms, coexisting CD must be suspected. Giardiasis caused by Giardia lamblia is one of the most common intestinal parasitic diseases. Giardia can be easily identified in duodenal biopsy specimen as a pear-shaped organism with two paired nuclei, located in lumen adjacent to the epithelium. A celiac serology may not be helpful as transient elevation of IgA-TTG and IgA-EMA levels may be seen in Giardia infection⁸². Repeating celiac serology after Giardia eradication may clarify the situation.
Diet and food allergy
Dietary proteins other than gluten can lead to malabsorption. The small intestinal histologic features may be like those seen in CD posing a diagnostic problem⁸³⁻⁸⁵. The condition is usually transient and clinical correlation with diet history is important.
MALNUTRITION
Patients with severe protein calorie malnutrition can present with diarrhea and malabsorption. The small bowel histologic features may be like those seen in CD with villous blunting, increased mucosal inflammation and sometimes increased IELs⁸⁶. However, these changes respond to oral or intravenous therapy⁸⁶.
MEDICATIONS
Certain medications can lead to malabsorption with small intestinal lesions akin to CD. Many types of NSAIDs show duodenal mucosal lesions including IELs⁸⁷. Olmesartan, an angiotensin II receptor blocker used in the treatment of CD can cause severe diarrhea. The duodenal biopsy shows changes mimicking CD with partial or complete villous atrophy and increased IELs⁸⁸. Other medications which can cause CD like intestinal changes include oxcarbazepine⁸⁹, mycophenolate mofetil⁹⁰, and azathioprine⁹¹. Immunotherapy used for cancer treatment can also lead to CD like enteropathy. History of medication usage, negative celiac serology and resolution of symptoms and intestinal pathologic features on discontinuation of medications are helpful features in arriving at a correct diagnosis Autoimmune enteropathy
In its pure form, autoimmune enteropathy is a disease of childhood, and it is likely that most pediatric cases have a genetic basis that remains to be uncovered. Approximately 20% of cases develop a celiac disease like pattern in the duodenum and is often associated with lymphocytosis throughout the remainder of the gastrointestinal tract⁹².
INTRACTABLE DIARRHEA OF INFANCY
Intractable diarrhea of early infancy is characterized by severe protracted diarrhea in young infants younger than 3 months of age. There is no apparent cause on detailed examination and hence no definite treatment can be started. The condition at times may be fatal⁹³. The condition is associated with small intestinal mucosal abnormalities such as total villous atrophy and crypt hyperplasia⁹⁴. Some cases may show villous atrophy associated with crypt hypoplasia⁹⁵⁻⁹⁷. Some of these cases might be based on autoimmunity and circulating antibodies to intestinal goblet cells and enterocytes have been demonstrated⁸⁹˒⁹⁰˒⁹⁷. A positive test for anti enterocyte antibody helps in establishing the diagnosis. A persistent high titre indicates a poor prognosis⁹⁷˒⁹⁸.
ADULT AUTOIMMUNE ENTEROPATHY
Adult form of autoimmune enteropathy associated with anti enterocyte antibodies may rarely present as refractory sprue⁹⁹˒¹⁰⁰. In one study the jejunal biopsies demonstrated subtotal villous atrophy and increased IELs before steroid treatment¹⁰⁰. Testing for anti enterocyte antibodies may be helpful in establishing diagnosis of autoimmune enteropathy.
CROHN’S DISEASE
Crohn’s disease can manifest as duodenitis, with villous atrophy and malabsorption. Diagnosis of Crohn’s disease remains a diagnosis of exclusion after ruling out CD by negative celiac serology, absence of HLA DQ2/DQ8, and lack of response to gluten free diet.
DIFFERENTIAL DIAGNOSIS OF LATENT CELIAC DISEASE
Intraepithelial lymphocytes with normal villous architecture (IELNVA) have been increasingly diagnosed in many patients with CD due to an increase in the utilization of serology testing and endoscopic examination of first degree relative of patients with CD. These findings although sensitive are not specific for CD and can be seen in a variety of other conditions. Several large studies have studied the prevalence of IELNVA. The prevalence of IELNVA varies from 1.3% to 2.2% when different cut off values are used¹⁰¹. Kakar et al. in their study, using 40 IELs as the cut off criteria for IEL, noted histologic pattern of IELNVA at a rate of 1.3% (43 out of 3190 patients undergoing upper GI endoscopy)⁸⁷. It is associated with CD (9.3%), immunologic disease (14.3%), NSAIDs use (14%), inflammatory bowel disease (11.6%), bacterial overgrowth (4.4%), tropical sprue (2.2%), microscopic colitis (6.9%), and other unspecified diagnosis (37.2%). Other diseases causing IELNVA include morbid obesity¹⁰², Helicobacter pylori gastritis⁶³˒¹⁰³˒¹⁰⁴, partially treated tropical sprue⁸⁷, cow’s milk protein intolerance⁶⁵. Immune disorders associated with IELNVA include Hashimoto’s thyroiditis, Graves disease, rheumatoid arthritis, psoriasis, multiple sclerosis, systemic lupus erythematosus, ankylosing spondylitis, progressive systemic sclerosis, and glomerulonephritis¹⁰⁵⁻¹⁰⁸. For many patients, a specific diagnosis is never established. The absence of HLA DQ2/DQ8 in at least half of these idiopathic cases excludes potential celiac disease¹⁰⁹.
Goldstein et al. suggested that increased density of IELs at the distal sides and tips of villi is a relatively specific and reproducible feature of latent or potential CD³⁶. Some other study, however, report no particular pattern of IEL distribution to be associated with latent or potential CD⁸⁷.
Idiopathic inflammatory bowel disease such as ulcerative colitis or Crohn’s disease and lymphocytic and collagenous colitis may be associated with IELNVA in the proximal small bowel⁸⁷˒¹¹⁰. HLA testing for relevant DQ3 alleles can be useful when diagnosis based on other tests is not useful.
Conclusion
There has been significant development in understanding the pathophysiology, pathogenesis, and diagnosis of celiac disease. The present review summarizes the important development in CD in last few years. The diagnostic criteria for the diagnosis have been developed in adult and pediatric population. There has also been refinement in celiac serology in the diagnosis of celiac disease. Despite these developments, histologic examination of small intestinal biopsies remains the gold standard for the diagnosis of celiac disease. Histologic examination of small intestine also helps to monitor the course of disease, response to treatment, and in detecting potential complications of celiac disease such as enteropathy associated T-cell lymphoma, adenocarcinoma, and concurrent intestinal diseases.
Conflict of Interest:
None
Funding Statement:
None.
Acknowledgements:
None.
References
1. Biagi F, Gobbi P, Marchese A, et al. Low incidence but poor prognosis of complicated coeliac disease: a retrospective multicentre study. Dig Liver Dis. 2014; 46:227–30.
2. Tinguria M. Rare Complications of Celiac Disease: Clinicopathologic Features, Medical Research Archives, [online] 2023: 11(4). https://doi.org/10.18103/mra.v11i4.3763
3. Tinguria M, Liaconis H. Cavitating mesenteric lymph node syndrome: A rare complication of celiac disease. A case report with clinicopathologic features and review of literature, Human Pathology Reports, Volume 26, 2021.
4. Shewry R, Tatham A, Kasarda D. Cereal proteins and coeliac disease. In: Marsh M, editor. Coeliac Disease. Oxford, UK: Blackwell Scientific Publications; 1992
5. Shewry P. What is gluten—why is it special? Front. Nutr. 2019, 6:101
6. Shan L, Molberg O, Parrot I, et al. Structural basis for gluten intolerance in celiac sprue. Science. 2002;297:2275–9.
7. Shan L, Qiao SW, Arentz-Hansen H, et al. Identification and analysis of multivalent proteolytically resistant peptides from gluten: implications for celiac sprue. J Proteome Res. 2005;4(5):1732–1741
8. Hemming-Harlo M, Lahdeaho M, Maki M. et al. Rotavirus vaccination does not increase type 1 diabetes and may decrease celiac disease in children and adolescents. Pediatr. Infect. Dis. J. 2019, 38:539– 41
9. Inns T, Fleming K, Iturriza-Gomara M. et al. Paediatric rotavirus vaccination, coeliac disease, and type 1 diabetes in children: a population-based cohort study. BMC Med. 2021, 19:147
10. Stordal K, Kahrs C, Tapia G. et al. Review article: exposure to microbes and risk of coeliac disease. Aliment Pharmacol Ther (2020) 53:43–62.
11. Nistico L, Fagnani C, Coto I, Percopo S, Cotichini R, et al. 2006. Concordance, disease progression, and heritability of coeliac disease in Italian twins. Gut 55:803–8
12. Kuja-Halkola R,Lebwohl B, Halfvarson J,Wijmenga C, Magnusson PK, Ludvigsson JF. 2016. Heritability of non-HLA genetics in coeliac disease: a population-based study in 107 000 twins. Gut 65:1793–98
13. Dube C, Rostom A, Sy R. et al. The prevalence of celiac disease in average-risk and at-risk Western European populations: a systematic review. Gastroenterology, 2005: 128:S57–S67
14. Sollid L, Markussen G, Ek J. et al. Evidence for a primary association of celiac disease to a particular HLA-DQ alpha/beta heterodimer. J Exp Med 169:345–350
15. Spurkland A, Ingvarsson G, Falk E. et al. Dermatitis herpetiformis and celiac disease are both primarily associated with the HLA-DQ (alpha 1*0501, beta1*02) or the HLA-DQ (alpha 1*03, beta 1*0302) heterodimers. Tissue Antigens, 1997 : 49 :29–34
16. Mearin M, Biemond I, Pena A. et al. HLA-DR phenotypes in Spanish coeliac children: their contribution to the understanding of the genetics of the disease. Gut, 1983: 24:532–537
17. Van de Wal Y, Kooy Y, Van V. et al. Selective deamidation by tissue transglutaminase strongly enhances gliadin-specific T cell reactivity. J Immunol, 1998a: 161:1585–1588
18. Hunt K, Zhernakova A, Turner G. et al. Newly identified genetic risk variants for celiac disease related to the immune response. Nat Genet, 2008: 40:395–402
19. Morbe U, Jorgensen P, Fenton T. et al. Human gut-associated lymphoid tissues (GALT); diversity, structure, and function. Mucosal Immunol. 2021: 14:793–802
20. Weisel N, Weisel F, Farber D. et al. Comprehensive analyses of B-cell compartments across the human body reveal novel subsets and a gut-resident memory phenotype. Blood, 2020: 136:2774–85
21. Bartolome-Casado R, Landsverk O, Chauhan S. et al. Resident memory CD8 T cells persist for years in human small intestine. J. Exp. Med. 2019: 216:2412–26
22. Bartolome-Casado R, Landsverk O, Chauhan S. et al. CD4+ T cells persist for years in the human small intestine and display a TH1 cytokine profile. Mucosal Immunol. 2021: 14:402–10
23. Landsverk O, Snir O, Casado R. et al. Antibody-secreting plasma cells persist for decades in human intestine. J. Exp. Med. 2017 : 214 :309–17
24. Mayassi T, Ladell K, Gudjonson H. et al. Chronic inflammation permanently reshapes tissue-resident immunity in celiac disease. Cell, 2019 : 176 :967–81. e19
25. Meresse B, Chen Z, Ciszewski C. et al. Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity 2004: 21:357–66
26. Hüe S, Mention J, Monteiro R. et al. A direct role for NKG2D/MICA interaction in villous atrophy during celiac disease. Immunity 2004: 21:367–77
27. Halstensen T, Brandtzaeg P. Activated T lymphocytes in the celiac lesion: non-proliferative activation (CD25) of CD4+ α/β cells in the lamina propria but proliferation (Ki-67) of α/β and γ/δ cells in the epithelium. Eur. J. Immunol. 1993: 23:505–10
28. Dahal-Koirala S, Risnes L, Sollid L. Pathogenesis of coeliac disease—a disorder driven by gluten-specific CD4+ T cells. In Coeliac Disease and Gluten-Related Disorders, ed. A Schiepatti, DS Sanders, 2022, pp. 41–68. London: Academic Press
29. Baklien K, Brandtzaeg P, Fausa O. 1977. Immunoglobulins in jejunal mucosa and serum from patients with adult coeliac disease. Scand. J. Gastroenterol. 1977: 12:149–59
30. Pinto D, Montani E, Bolli M. et al. A functional BCR in human IgA and IgM plasma cells. Blood 2013: 121:4110–14
31. Di Niro R, Mesin L, Raki M. et al. Rapid generation of rotavirus specific human monoclonal antibodies from small-intestinal mucosa. J. Immunol. 2010: 185:5377–83
32. Hoydahl L, Richter L, Frick R. et al. Plasma cells are the most abundant gluten peptide MHC-expressing cells in inflamed intestinal tissues from patients with celiac disease. Gastroenterology 2019: 156:1428–39.e10
33. Diosdado B, Van Bakel H, Strengman E. et al. Neutrophil recruitment and barrier impairment in celiac disease: a genomic study. Clin. Gastroenterol. 2007: Hepatol. 5:574–81
34. Moran C, Kolman O, Russell G. et al. Neutrophilic infiltration in gluten-sensitive enteropathy is neither uncommon nor insignificant: assessment of duodenal biopsies from 267 pediatric and adult patients. Am. J. Surg. Pathol. 2012: 36:1339–45
35. Tokoyoda K, Hauser A, Nakayama T. et al. Organization of immunological memory by bone marrow stroma. Nat. Rev. Immunol. 2010: 10:193–200
36. Landsverk O, Snir O, Casado R. et al. Antibody-secreting plasma cells persist for decades in human intestine. J. Exp. Med. 2017: 214:309–17
37. Nilsen E, Lundin K, Krajci P. et al. Gluten specific, HLA-DQ restricted T cells from coeliac mucosa produce cytokines with Th1 or Th0 profile dominated by interferon gamma. Gut 1995: 37:766–76
38. 35. Dwinell M, Lugering N, Eckmann L. et al. Regulated production of interferon-inducible T-cell chemoattractants by human intestinal epithelial cells. Gastroenterology 2001: 120:49–59
39. Mention J, Ben Ahmed M, Begue B. et al. Interleukin 15: a key to disrupted intraepithelial lymphocyte homeostasis and lymphomagenesis in celiac disease. Gastroenterology (2003): 125:730 – 45
40. Waldmann T. The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nat. Rev. Immunol. 2006: 6:595–601
41. Jabri B, Abadie V. IL-15 functions as a danger signal to regulate tissue-resident T cells and tissue destruction. Nat. Rev. Immunol. 2015:15:771–83
42. Molberg O, McAdam S, Korner R. et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat. Med. 1998: 4:713–17
43. Van de Wal Y, Kooy Y, Van V. et al. Selective deamidation by tissue transglutaminase strongly enhances gliadin-specific T cell reactivity. J Immunol, 1998a: 161:1585–1588
44. Lorand L, Graham R. Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat. Rev. Mol. Cell Biol. 2003: 4:140–56
45. Siegel M, Strnad P, Watts R. et al. Extracellular transglutaminase 2 is catalytically inactive but is transiently activated upon tissue injury. PLOS ONE, 2008, 3:e1861
46. Tjon J, Kooy-Winkelaar Y, Tack G. et al. DNAM-1 mediates epithelial cell-specific cytotoxicity of aberrant intraepithelial lymphocyte lines from refractory celiac disease type II patients. J Immunol. (2011) 186:6304–12. doi : 10.4049/j immunol.
47. Jabri B, Ebert E. Human CD8+ intraepithelial lymphocytes: a unique model to study the regulation of effector cytotoxic T lymphocytes in tissue. Immunol Rev. 2007 Feb: 215:202-14.
48. Bauer S, Groh V, Wu J. et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science (1999): 285:727–729
49. Stene L, Honeyman M, Hoffenberg E. et al. Rotavirus infection frequency and risk of celiac disease autoimmunity in early childhood: a longitudinal study. Am J Gastroenterol (2006): 101:2333–2340
50. Brocchi E, Corazza G, Galetti G. et al. Endoscopic demonstration of loss of duodenal folds in the diagnosis of celiac disease. N Engl J Med. 1988; 319:741–744.
51. Maurino E, Capizzano H, Niveloni S. et al. Value of endoscopic markers in celiac disease. Dig Dis Sci. 1993 ;38 : 2028–2033.
52. Sanfilippo G, Pantane R, Fusto A. et al. Endoscopic approach to childhood celiac disease. Acta Gastroenterologica. 1986;49:401–408.
53. Holmes R, Hourihane D, Booth C. Dissecting microscope appearance of jejunal biopsy specimen from patients with “idiopathic steatorrhea”. Lancet. 1961;277:81–83.
54. Stewart J, Pollock D, Hoffbrand A. et al. A study of proximal and distal intestinal structure and absorptive function in idiopathic steatorrhea. Q J Med. 1967;36:425–444.
55. Rostom A, Murray J, Kagnoff M. American Gastroenterological Association (AGA) institute technical review on the diagnosis and management of coeliac disease. Gastroenterology. 2006;131:1981–2002
56. Mee A, Burke M, Vallon A. et al. Small bowel biopsy for malabsorption: comparison of the diagnosis adequacy of endoscopic forceps and capsule biopsy specimens. Br Med J. 1985;291:769–772.
57. Lindfors K, Koshinen O, Kaukinen K. An update on the diagnostics of celiac disease. Int Rev Immunol. 2011;30:185–196.
58. Pelkowski T, Viera A. Celiac disease: diagnosis and management. Am Fam Physician. 2014 ;89:99–105.
59. Ludvigsson J, Bai J, Biagi F. et al. Diagnosis and management of adult coeliac disease: guidelines from the British Society of Gastroenterology. Gut. 2014;63:1210–1228
60. Ensari A. Gluten-sensitive enteropathy (celiac disease): controversies in diagnosis and classification. Arch Pathol Lab Med. 2010;134:826–836.
61. Lebwohl B, Kapel R, Neugut A. et al. Adherence to biopsy guidelines increases celiac disease diagnosis. Gastrointest Endosc. 2011 ;74 :103–109.
62. Wallach T, Genta R, Lebwohl B. et al. Adherence to celiac disease and eosinophilic esophagitis biopsy guidelines is poor in children. J Pediatr Gastroenterol Nutr. 2017;65:64–68.
63. Goldstein N. Proximal small-bowel mucosal villous intraepithelial lymphocytes. Histopathology. 2004;44:199–205.
64. Ferguson A, Murray D. Quantitation of intraepithelial lymphocytes in human jejunum. Gut. 1971;12:988–994.
65. Phillips A, Rice S, France N. et al. Small intestinal intraepithelial lymphocyte levels in cow’s milk protein intolerance. Gut. 1979;20:509–512
66. Oberhuber G, Granditsch G, Volgelsang H. The histopathology of coeliac disease: time for a standardized report scheme for pathologists. Eur J Gastroenterol Hepatol. 1999;11: 1185–1194.
67. Marsh MN, Johnson MW, Rostami K. Mucosal histopathology in celiac disease: a rebuttal of Oberhuber’s sub-division of Marsh III. Gastroenterol. Hepatol. Bed Bench 2015; 8; 99–109
68. Corazza G, Villanacci V, Zambelli C, et al. Comparison of the interobserver reproducibility with different histologic criteria used in celiac disease. Clin Gastronterol Hepatol. 2007;5:838–843
69. Husby S, Murray J, Katzka D. AGA Clinical Practice Update on Diagnosis and Monitoring of Celiac Disease— changing utility of serology and histologic measures: expert review. Gastroenterology 2019; 156; 885–889.
70. Brown I. Bettington M, Rosty C. The role of histopathology in the diagnosis and management of coeliac disease and other malabsorptive conditions. Histopathology.2021 Jan;78(1):88-105.
71. Adelman D, Murray J, Wu T. et al. Measuring change in small intestinal histology in patients with celiac disease. Am. J. Gastroenterol. 2018; 113; 339–347.
72. Hære P, Høie O, Schulz T. et al. Long-term mucosal recovery and healing in celiac disease is the rule not the exception. Scand. J. Gastroenterol. 2016; 51;1439–1446.
73. Ochs H, Ament M, Davis S. Giardiasis with malabsorption in X-linked agammaglobulinemia. N Engl J Med. 1972;287:341–342.
74. Rhee J, Gryboski J, Sheahan D. et al. Reversible enteritis and lymphopenia in infantile x-linked agammaglobulinemia. Am J Dig Dis. 1975;20: 1071–1075.
75. Malamut G, Verkarre V, Suarez F. et al. The enteropathy associated with common variable immunodeficiency: the delineated frontier with celiac disease. Am J Gastroenterol. 2010;105: 2262–2275.
76. Joo M, Sang H, Sun H. et al. Nodular lymphoid hyperplasia and histologic changes mimicking celiac disease, collagenous sprue, and lymphocytic colitis in a patient with selective IgA deficiency. Pathol Res Pract. 2009;205:876–880.
77. Perlmutter D, Leichtner A, Goldman H. et al. Chronic diarrhea associated with hypogammaglo bulinemia and enteropathy in infants and children. Dig Dis Sci. 1985;30:1149–1155.
78. Horowitz A, Lorenzsonn V, Olsen W. et al. Small intestinal disease in T cell deficiency. J Pediatr. 1974;85: 457–462.
79. Sharma P, Baloda V, Gahlot GP. et al. Clinical, endoscopic, and histological differentiation between celiac disease and tropical sprue: A systematic review. J Gastroenterol Hepatol. 2019;34:74–83.
80. Troeger H, Loddenkemper C, Schneider T. et al. Structural and functional changes of the duodenum in human noro1070 virus infection. Gut 2009; 58; 1070–1077.
81. Wolf J, Schreiber D. Viral gastroenteritis. Med. Clin. North Am. 1982; 66; 575–595.
82. Hanevik K, Wik E, Langeland N. et al. Transient elevation of anti-transglutaminase and anti-endomysium antibodies in Giardia infection. Scand J Gastroenterol. 2018 ;53 :809–812.
83. Rosekrans P, Meijer C, Cornelisse C. et al. Use of morphometry and immunohistochemistry of small intestinal biopsy specimens in the diagnosis of food allergy. J Clin Pathol. 1980; 33:125–130.
84. Vitoria J, Camarero C, Sojo A. et al. Enteropathy related to fish, rice, and chicken. Arch Dis Child. 1982;57:44–48.
85. Challacombe D, Wheeler E, Campbell P. Morphometric studies and eosinophil cell counts in the duodenal mucosa of children with chronic nonspecific diarrhoea and cow’s mild allergy. J Pediatr Gastroenterol Nutr. 1986; 5:887–891.
86. Shiner M, Redmond A, Hansen J. The jejunal mucosa in protein-energy malnutrition. A clinical, histological, and ultrastructural study. Exp Mol Pathol. 1973;19:61–78.
87. Kakar S, Nehra V, Murray J. et al. Significance of intraepithelial lymphocytosis in small bowel biopsy samples with normal mucosal architecture. Am J Gastroenterol. 2003; 98:2027–2033.
88. Sáez González E, Díaz Jaime F, Del Val Antoñana A. Clinical, laboratory, serological, and histological profile of sprue-like enteropathy associated with olmesartan use. Rev Esp Enferm Dig. 2016; 108:685–686.
89. Gonzálezcordero P, Fernandezgonzalez N, Molinainfante J. Sprue-Like Enteropathy Associated With Oxcarbazepine. Am J Gastroenterol. 2016; 111:1662–1663.
90. Parfitt J, Jayakumar S, Driman D. Mycophenolate mofetil-related gastrointestinal mucosal injury: variable injury patterns, including graft-versus-host disease-like changes. Am J Surg Pathol. 2008 ;32 :1367–1372.
91. Ziegler T, Fernández-Estívariz C, Gu L. et al. Severe villus atrophy and chronic malabsorption induced by azathioprine. Gastroenterology. 2003; 124:1950–1957.
92. Masia R, Peyton S, Lauwers G. et al. Gastrointestinal biopsy findings of autoimmune enteropathy: a review of 25 cases. Am. J. Surg. Pathol. 2014; 38; 1319–1329.
93. Avery G, Villavicencio O, Lilly J. et al. Intractable diarrhea in early infancy. Pediatrics. 1968 ;41 :712–722.
94. Rossi T, Lebenthal E, Nord K. et al. Extent and duration of small intestinal mucosal injury in intractable diarrhea of infancy. Pediatrics. 1980; 66:730–735.
95. Davidson G, Cutz E, Hamilton J. et al. Familial enteropathy: a syndrome of protracted diarrhea from birth, failure to thrive, and hypoplastic villous atrophy. Gastroenterology. 1978;75:783–790.
96. Fisher S, Boyle J, Holtzapple P. Chronic protracted diarrhea and jejunal atrophy in infant: cimetidine-associated stimulation of jejunal mucosal growth. Dig Dis Sci. 1981 ;26 :181–186.
97. Savage M, Mirakian R, Wozniak E. et al. Specific autoantibodies to gut epithelium in two infants with severe protracted diarrhea. J Pediatr Gastroenterol Nutr. 1985;4:187–195.
98. Mirakian R, Richardson A, Milla P. et al. Protracted diarrhea of infancy: evidence in support of an autoimmune variant. Br Med J. 1986; 293:1132–1136.
99. Ryan B, Kelleher D. Refractory celiac disease. Gastroenterology. 2000; 119:243–251
100. Corazza G, Biagi F, Volta U. et al. Autoimmune enteropathy and villous atrophy in adults. Lancet. 1997 ;350 :106–109.
101. Mahadeva S, Wyatt J, Howdle P. Is a raised intraepithelial lymphocyte count with normal duodenal villous architecture clinically relevant? J Clin Pathol. 2002;55:424–428.
102. Harpaz N, Levi G, Yurovitsky A, et al. Intraepithelial lymphocytosis in architecturally normal small intestinal mucosa: association with morbid obesity. Arch Pathol Lab Med. 2007; 131:344.
103. Nahon S, Patey-Mariaud De Serre N, Lejeune O. et al. Duodenal intraepithelial lymphocytosis during Helicobacter pylori infection is reduced by antibiotic treatment. Histopathology. 2006;48: 417–423
104. Memeo L, Jhang J, Hibshoosh H, et al. Duodenal intraepithelial lymphocytosis with normal villous architecture
105. Ferguson A. Intraepithelial lymphocytes of the small bowel. Gut. 1977;18:921–937.
106. Michaelsson G, Kraaz W, Gerden B. et al. Increased lymphocytic infiltration in duodenal mucosa from patients with psoriasis and serum IgA antibodies in gliadin. Br J Dermatol. 1995; 133:896–904.
107. Brown I, Mino-Kenudson M, Deshpande V. et al. Intraepithelial lymphocytosis in architecturally preserved proximal small intestinal mucosa. Arch Pathol Lab Med. 2006;130:1020–1025
108. Rostoker G, Delchier J, Chaumette M. Increased intraepithelial T lymphocytes in primary glomerulonephritis: a role of oral tolerance breakdown in the pathophysiology of human primary glomerulonephritis. Nephrol Dial Transpant. 2001;16:513–517.
109. Vande Voort J, Murray J, Lahr B. et al. Lymphocytic duodenosis and the spectrum of celiac disease. Am. J. Gastroenterol. 2009; 104; 142–148.
110. Tobin J, Sinha B, Ramani P. et al. Upper gastrointestinal mucosal disease in pediatric Crohn disease and ulcerative colitis: a blinded, controlled study. J Pedistr Gastroenterol Nutr. 2001;32:443–448.