






Primary Vitreoretinal Lymphoma: Diagnosis & Management
PRIMARY VITREORETINAL LYMPHOMA: A REVIEW ON DIAGNOSIS AND MANAGEMENT CHALLENGES
Meera Khosla, MD1, Noah G. Finkel, MD2, Faizan Aijaz, MD3, Juliana Rios, MD4, Amy Scheffler, MD5, Bin S. Teh, MD6, Isai Raval Pingali, MD7, Ethan B. Burns, MD8
- Department of Internal Medicine, Houston Methodist Hospital, Houston, TX
- Ocular Oncology and Vitreoretinal Surgery, Retina Consultants of Texas, Houston, TX
- Department of Radiation Oncology, Houston Methodist Hospital, Houston, TX
- Houston Methodist Neel Cancer Center, Houston Methodist Hospital, Houston, TX
- Department of Internal Medicine, Houston Methodist Hospital, Houston, TX
- Department of Radiation Oncology, Houston Methodist Hospital, Houston, TX
- Department of Internal Medicine, Houston Methodist Hospital, Houston, TX
- Department of Internal Medicine, Houston Methodist Hospital, Houston, TX
OPEN ACCESS
PUBLISHED: 30 December 2024
CITATION: KHOSLA, Meera et al. PRIMARY VITREORETINAL LYMPHOMA: A REVIEW ON DIAGNOSIS AND MANAGEMENT CHALLENGES. Medical Research Archives, [S.l.], v. 12, n. 12, jan. 2025. Available at: <https://esmed.org/MRA/mra/article/view/6142>.
COPYRIGHT: © 2025 European Society of Medicine. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
ISSN 2375-1924
Abstract
Primary vitreoretinal lymphoma (PVRL) is a rare type of primary central nervous system lymphoma (PCNSL), with disease localized to retina or vitreous humor, not to be confused with secondary causes of systemic lymphoma which metastasizes from a non-ocular primary site and frequently has disease dissemination to the uvea or choroid. Diffuse large B-cell lymphoma (DLBCL) is the most commonly diagnosed histologic subtype, with a minority of cases diagnosed as T-cell lymphoma. The diagnosis is frequently delayed and often mistaken for uveitis and treated with immunosuppressive therapies before a diagnosis of PVRL is made. This review aims to summarize the current understanding of PVRL, including its pathogenesis, diagnostic challenges, and management strategies.
Keywords
primary vitreoretinal lymphoma, diagnosis, management, diffuse large B-cell lymphoma, ocular oncology
Introduction
PVRL is an aggressive malignancy that comprises less than 1% of non-Hodgkin lymphoma (NHL) cases. It is a rare subtype of primary central nervous system lymphoma (PCNSL) with disease localized to retina or vitreous humor, not to be confused with secondary causes of systemic lymphoma which metastasizes from a non-ocular primary site and frequently has disease dissemination to the uvea or choroid. Diffuse large B-cell lymphoma (DLBCL) is the most commonly diagnosed histologic subtype, with a minority of cases diagnosed as T-cell lymphoma. The diagnosis is frequently delayed and often mistaken for uveitis and treated with immunosuppressive therapies before a diagnosis of PVRL is made.
Primary vitreoretinal lymphoma carries a poor prognosis once central nervous system (CNS) metastasis occurs, and whether currently available therapeutic modalities prevents CNS dissemination is controversial. Overall survival (OS) ranges from 58 to 75 months and median progression-free survival is between 18 to 29 months. Given the rarity and fragility of lymphoma cells in the vitreous, establishing a diagnosis is challenging and requires an ocular oncologist with expertise in performing a vitrectomy with direct visualization of malignant cells.
There is a paucity of standardized guidelines regarding the management of this disease and it is often institution specific. Treatment for PVRL localized to the eye is typically local therapy with either intravitreal injections with methotrexate and/or rituximab or low dose orbital radiation, with systemic therapies reserved for bilateral ocular involvement, CNS dissemination, or locally recurrent disease. However, prospective studies are necessary to establish optimal treatment strategies in the management of this rare disease. This review aims to focus on the history, epidemiology, diagnostic methods, and treatment challenges for patients with PVRL.
Nomenclature and Classification
Originally known as ocular reticulum cell sarcoma and later intraocular lymphoma (IOL), PVRL was aptly renamed due to its association with vitreoretinal infiltrates. IOLs are categorized into two subtypes: retinal lymphoma and uveal lymphoma. Retinal lymphomas can be further subdivided into vitreal, vitreoretinal, and retinal lymphomas, with these distinctions determined by ophthalmoscopy. Uveal lymphomas involve the iris, ciliary body, and choroid and are typically seen with systemic lymphomas that have subsequently metastasized to the region, rather than a primary ocular neoplasm. Retinal lymphomas are often low-grade extranodal marginal zone lymphoma (EMZL), and can be differentiated from PVRL by fluorescence in situ hybridization (FISH) identification of MALT1 and BCL10, or classification of mucosa-associated lymphoid tissue (MALT). PVRL is a retinal lymphoma that involves the vitreous body, retina, and optic nerve. It is important to note that vitreoretinal lymphoma can also be caused by ocular involvement of PCNSL or secondary disease from systemic DLBCL.
Epidemiology
PVRL is most often diagnosed in the fifth to sixth decade of life. It is very rare to see cases in adolescence, and when they do occur, they are associated with human immunodeficiency virus (HIV), or other immunocompromised states. The most well-documented risk factors for PVRL include Epstein-Barr virus (EBV) and HIV. Primary intraocular lymphomas (PIOL) comprise 1.86% of ocular malignancies and 1-2% of extranodal lymphomas. While 15-25% of PCNSL patients develop ophthalmic manifestations of lymphoma, 56-90% of PIOL patients have or will develop CNS manifestations of lymphoma.
Historically, incidence in the United States has been cataloged by the Central Brain Tumor Registry of the United States (CBTRUS). From 2015–2019 the CBTRUS reported that the annual age-adjusted incidence rate of all brain tumors was 24 per 100,000 population, with an estimated 93,000 new cases diagnosed each year, 29% of which were expected to be malignant. Based on a 2006 study by Chan et al., there are an estimated 2,000 cases of PCNSL per year.
Approximately 400 were PVRL. A retrospective review from British Columbia spanning 1990 to 2010 found 22 cases of PVRL, resulted in an estimated incidence of 0.047 cases per 100,000 or approximately 1 case per 2 million patients. There is no racial predilection, and females tend to have a higher incidence than males.
Pathogenesis
The mechanism of PVRL lymphomagenesis is controversial, but two prevailing theories suggest PVRL arises from aberrant cytokine signaling, and another suggests a virus-induced immunogenic trigger.
The first theory: abnormal cytokine/cytokine receptor signaling, which augments leukocyte trafficking, migration, and activation. Two pairs of receptors and ligands have been identified: CXCR4 and its ligand stromal cell-derived factor (SDF-1) also called CXCL12, and CXCR5 which binds to B-lymphocyte chemoattractant (BCL-1), also called CXCL13. Lymphoma cells have increased expression of CXCR4 and CXCR5. Conversely, SDF-1 and BCL-1 have increased expression on normal retinal pigment epithelium (RPE). Falkenhagen et al. theorized that expression of these chemokines within the intraocular compartment may contribute to malignant lymphoma cells traveling to the retinal pigment epithelium. This pairing of chemokines and chemokine receptors is being studied as a potential target for inhibitors in treatment of PIOL.
The second theory of pathogenesis involves an extrinsic immune trigger altering lymphocyte production. Epstein-Barr virus (EBV) has a well-known association with PCNSL in the setting of acquired immunodeficiency syndrome (AIDS), where proliferation of lymphocytes is unchecked by T-suppressor lymphocytes causing increased proliferation and increased risk for aberrancy. Epstein-Barr virus has been linked to Burkitt, Hodgkin, and DLBCL. EBV is believed to result in DLBCL progenitor cells arrested in germinal center transit, a crucial moment for primary intraocular lymphomagenesis.
Both the chemokine pathway and infectious pathway take advantage of the B-cell proliferation mechanism. Normally, TH1 lymphocytes create humoral immunity against pathogens and stimulate production of antibody-secreting plasma cells and B-cells. These B-cells then differentiate and migrate into the dark zone of the germinal center, where they undergo somatic hypermutation, then migrate to the light zone, where they undergo class switching. B-cells that remain in the germinal center and do not undergo differentiation lack genetic control and are susceptible to monoclonal expansion. Based on genomic sequencing, most PCNSL cellular expression does not differ from DLBCL, however some heterogeneity exists.
The exact origin of neoplastic cells in PVRL remains unclear. The eye is an immune-privileged site with low expression of major histocompatibility complex (MHC) and higher expression of immunosuppressive molecules such as transforming growth factor (TGF-β), macrophage migration inhibitory factor, and Fas ligands, which collectively impair T-helper 1 (Th1)-mediated signaling processes and thus antitumor responses. Like PCNSL, it is hypothesized that PVRL originates outside the CNS and eventually evades the immune system, growing uninhibited in the immunosuppressive ocular environment. A specific signaling molecule has not been identified that directs neoplastic cells into the brain. Deeper insights into the driving factors that localize the neoplastic cells to the eye and features which enables penetration into the blood-brain-barrier (BBB) and blood-retinal-barrier (BRB) are needed.
PVRL has identified many mutations in immunoglobulin (IG) genes. IGHV4-34 gene sequence is seen at a considerably higher frequency than other lymphomas including PCNSL and activated B-cell (ABC) DLBCL. This restricted IG repertoire lends support to an underlying antigen selection process of PVRL tumors, although the site in which these mutations are acquired has yet to be determined. Montesinos-Rongen et al. showed that all IGHV4-34-mutant antibodies recognize the Galectin-3 antigen, which is expressed by cells in the microenvironment of the brain. Galectin-3 interacts with retinal pigment epithelial cells and may be an important target antigen of PVRL. The tumor microenvironment is considered to have a significant influence over survival and proliferation of neoplastic cells. An elevated level of interleukin-10 (IL-10) has been observed in the vitreous or aqueous humor in lymphomatous eyes and is considered to have diagnostic value. Touitou et al. demonstrated minimal change in the levels of IL-10 in lymphomatous eyes with T-cell stimulation, suggesting an intrinsic production by tumor cells. IL-10 is an anti-inflammatory cytokine which facilitates tumor cell survival and also is a growth factor for B-cells. Furthermore, additional cytokines secreted by T helper 2 (Th2) cells were not identified in the vitreous, similarly suggesting a limited role of infiltrating Th1 and Th2 cells. Further studies are necessary to identify treatment interventions that can re-engage the anti-tumor response in this sanctuary site.Clinical Findings
Symptoms are frequently vague, and include blurry vision in 40–50%, decreased visual acuity in 25–30%, and “floaters” in 20–25% of cases. The paucity of specific symptoms results in misdiagnosed ocular conditions, such as posterior vitreous detachment and uveitis. The rarity and vague symptoms associated with PVRL may delay the diagnosis by 6–40 months from the time of symptoms onset. In addition, PCNSL may precede or follow PVRL, and presenting symptoms may include a spectrum of neurological changes including mental status changes, cognitive decline, vision changes, headaches, cranial nerve deficits, hemiplegia/hemiparesis, aphasia, ataxia, and seizures. Concomitant CNS involvement is present in 16–34% of patients at the time of diagnosis of PVRL. Subsequent CNS dissemination occurs in 42–92% of patients with PVRL with a mean time to metastasis ranging from 8 to 29 months. Silverman and colleagues reported upon 65 patients with PVRL, in which positive cerebrospinal fluid (CSF) was identified in 16.9% of patients without radiographic evidence of disease on magnetic resonance imaging (MRI). With frequent symptoms overlapping with other ocular conditions, clinicians must have a high index of suspicion, particularly in the absence of neurologic findings. The disease occurs in both eyes in 64–83% of patients at presentation and may often present with asymmetric severity.Diagnostic Approach
Diagnosing PVRL in a timely manner remains a challenge, as symptoms are often insidious and non-specific. The median time from symptom onset to diagnosis of PVRL ranges from 6–40 months, whereas the median time from symptoms to diagnosis of PCNSL is approximately 35 days. Definitive diagnosis of PVRL requires a vitrectomy with cytopathologic analysis. Suspicion for PVRL should be raised for large inflammatory cells in the vitreous without conjunctival injection or pain in an older patient; unexplained visual changes such as non-resolving vitritis; subretinal or sub-RPE white deposits plus vitreous cells; or neurologic features that might suggest disseminated CNS involvement.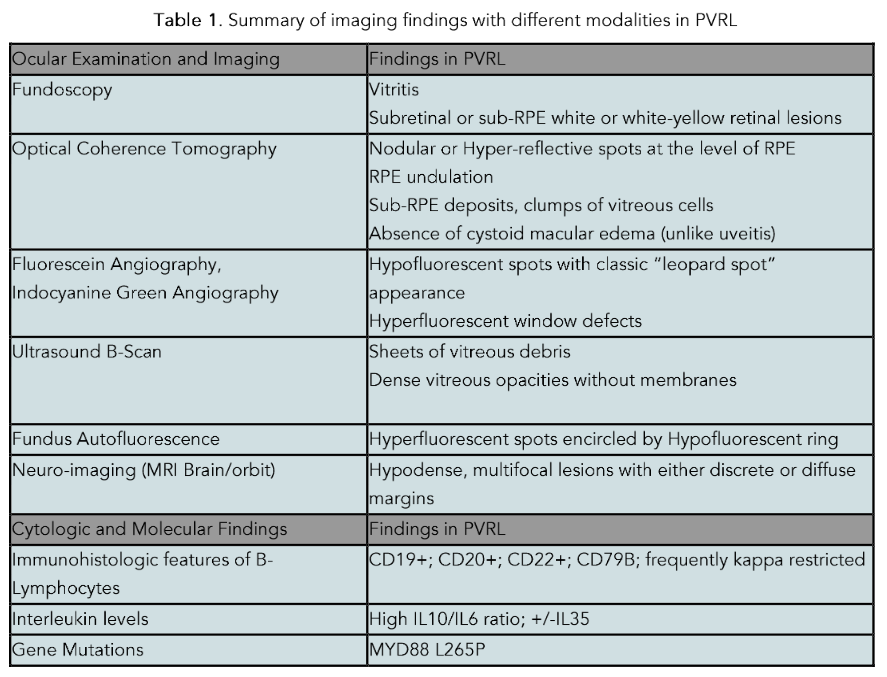
Neuroimaging
If the first symptom is exclusively ocular in nature, evaluation with a retina specialist or ocular oncologist is recommended. Neuroimaging also plays an important role in assessing the extent of disease. Positron emission tomography (PET) scans are often utilized in resource abundant medical centers to assess for extra-CNS sites of disease which occur in approximately 10% or fewer of cases.
Optical Coherence Tomography
Optical coherence tomography (OCT) is one non-invasive method of diagnosing and monitoring PVRL which can provide highly detailed, cross-sectional, two-dimensional images of the macula, the functional center of the retina. It enables the ophthalmologist to identify subretinal or sub-retinal pigment epithelial deposits which represent lymphomatous deposits. Large central clumps of cells can be seen in a so-called pseudovitelliform configuration under the fovea, the center of the macula. OCT is not definitive, and definitive cytopathology must be performed from a vitrectomy specimen to establish the diagnosis.
Ophthalmic Ultrasound
Ophthalmic ultrasound (B-scan) is a low cost and noninvasive method of analyzing the vitreous and retina and is commonly used by ocular oncologists to detect retinal detachments and mass lesions. Specifically for vitreoretinal lymphoma, B-scan in the hands of an experienced ultrasonographer can be used with very high sensitivity to identify vitreous cells suspicious for PVRL and indicate the need for a vitrectomy.
Biopsy
Direct biopsy of malignant cells is required for definitive diagnosis for lymphoma, and samples can be obtained from vitrectomy, cerebrospinal fluid, or primary CNS lesions. Vitrectomy for vitreous aspirate is obtained typically after there is clinical suspicion for PVRL and/or if neuroimaging and CSF analysis is negative. In cases in which subretinal or sub-RPE deposits are present with minimal vitreous involvement, retinal biopsy can be performed.
Histology and Cytology
On histological evaluation, PVRL is characterized by perivascular or subretinal infiltration, pleomorphic cells, indented or folded nuclei, minimal basophilic cytoplasm, and are often seen in a necrotic cellular background. Mitotic figures and Ki-67 index frequently suggest high rate of cellular proliferation. Cytology alone has a 45%–60% sensitivity in diagnosing PVRL. Flow cytometry is useful in establishing monoclonality and delineating from other conditions, as an elevated or depressed kappa:lambda light chain ratio favors monoclonality, whereas a normal ratio may be more in favor of inflammatory conditions such as uveitis.
Immunohistochemistry and flow cytometry often show positive B-lymphocyte expression of CD20, CD79a, PAX5, BCL-2, MUM1, and BCL-6. In one retrospective study, 84 vitrectomy, chorioretinal, and enucleated samples from patients that had clinically suspected primary intraocular lymphoma or chronic idiopathic uveitis were histologically analyzed. Of this group, 62 patients were diagnosed as reactive cellular infiltrate, and 17 patients (20%) were diagnosed as either suspicious of neoplastic disease or as definite malignant lymphoma with immunohistochemistry staining positive for CD79+, CD20+, BCL-2+, BCL-6+, MUM1+, and monotypical expression for IgM+.
Primary vitreoretinal lymphoma is frequently associated with MYD88 and/or CD79B mutations. MYD88 L265P was observed in 88% of vitreous samples in 25 patients with PVRL. CD79B motif mutations were observed in 35% of vitreous samples reported by Soussain et al., but were associated with more aggressive disease and CNS involvement. Other mutations such as BCL6, PIM1, PAX5, RHOH, MYC, BTG2, KLHL14, SUSD2 or chromosomal mutations (18q21 gain, or loss of 9p21, 8q12, and 6q21) have been reported in association with PVRL.
Cytokine Levels
Increased levels of IL-10 have been shown to favor the diagnosis of PVRL, whereas elevated levels of IL-6 are associated with inflammatory or reactive conditions. The ratio of IL-10/IL-6 may be useful to delineate the diagnosis in situations where cytologic and flow cytometry are not clear. Increased IL-10/IL-6 ratios favor PVRL, whereas low IL-10/IL-6 ratios are neither sensitive nor specific. Cassoux et al. described an IL-10 cutoff of 50 pg/mL in the aqueous humor (89% sensitivity, 93% specificity). More recently, Costopoulos et al. proposed a new score, called the Interleukin Score for Intra-Ocular Lymphoma (ISOLD). This score calculates the IL-10 and IL-6 concentrations in two distinct formulas, one for aqueous humor (via aqueous tap) and one for vitreous humor. The score from the formulas predicts the probability of having PVRL with a high sensitivity (93%) and specificity (95%) and has been validated as a clinical tool for diagnosing PVRL.
Treatment
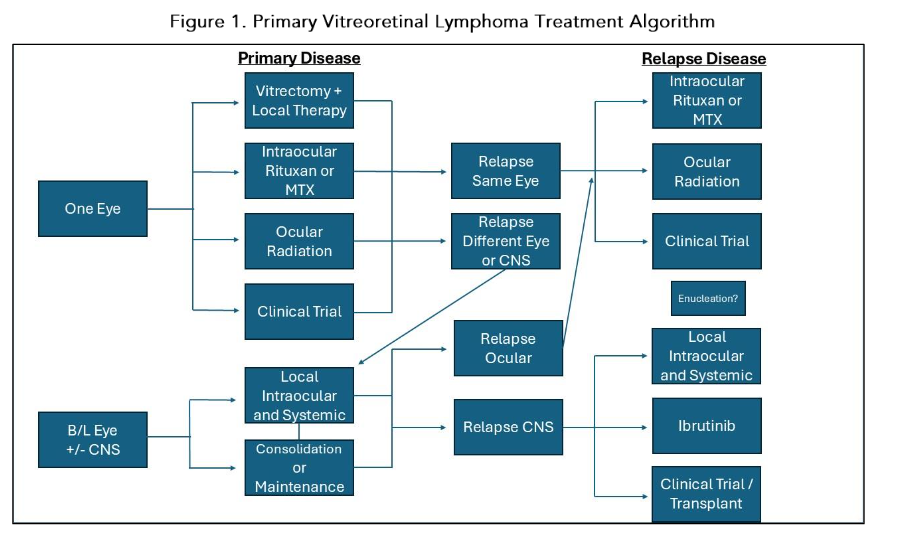
Given the rarity of this disease, there is no established standard of care, with some suggested considerations in Figure 1 based on EHA-ESMO 2024 guidelines and expert consensus. Effective management requires a multidisciplinary collaboration between ocular oncology, radiation oncology, and malignant hematology/neuro-oncology. PVRL is sensitive to both chemotherapy and radiation, but relapse rates are high. Treatment modalities for PVRL have evolved to include intraocular chemotherapy for localized disease, systemic chemotherapy with diverse consolidation strategies including autoHCT, CAR-T therapy, whole-brain radiotherapy (WBRT), or a combination strategy depending on the extent of the disease and patient factors. Selection of an appropriate treatment modality is contingent upon whether patients have localized PVRL or CNS involvement (Figure 1).
Ocular Therapy
METHOTREXATE
Intravitreal (IV) injection of methotrexate (MTX) is an effective therapy for unilateral or bilateral PVR. Whether IV MTX prevents CNS relapse remains a controversial topic. The largest single center study on the use of IV methotrexate was published by Habot-Wilner et al. A retrospective review of 81 patients with PVRL (134 eyes) received a mean (±SD) of 19 (±7) injections; however, only 5 (±4) injections were needed to reach complete remission. Local recurrence occurred in two of the 81 patients. Overall, 80.2% of eyes had an initial moderate-severe visual loss, and >50% of them improved. Reversible keratopathy was the most prevalent side-effect. A total of 18.5% developed intraocular pressure (IOP) elevation due to angle neovascularization after 16 injections, which could be reversed with prompt IV injection of bevacizumab.
The protocol included a total of 25 injections of 400 microg/0.1 ml MTX IV dispersed twice weekly for 4 weeks, once weekly for 8 weeks, and then once monthly for 9 months. Common complications from IV MTX include corneal epitheliopathy, vitreous hemorrhage, keratopathy, cataract formation, optic atrophy, and conjunctival injection. Current iterations of this protocol at many centers involve less frequent injections with a total volume injected of 0.05 ml rather than 0.1 ml with comparable efficacy results.
RITUXIMAB
IV rituximab is another option for localized PVRL management. Rituximab is an anti-CD20 monoclonal antibody commonly used to treat B cell lymphomas. IV rituximab injections can be used as monotherapy, in conjunction with MTX, or as maintenance therapy. Rituximab monotherapy has been effective in managing PVRL, which was reported in a retrospective study treating 12 eyes in 7 patients with complete response (CR) at a mean of 8.16 ± 4.62 months.
In a retrospective study of 48 eyes in 34 patients, 37% were treated with rituximab alone and the rest used it in combination with IV MTX. Results showed 64.6% achieved complete remission after a median of 3 injections, 22.9% had partial remission and 8% had no response. A prospective study using rituximab in twenty eyes in 13 patients previously treated with MTX discontinued due to complications revealed PVRL disease control and reduced retinal invasion and decreased number of keratic precipitates (KP). This study went on to report that CNS dissemination occurred in 69% of patients. The usual dosage of rituximab is 1 mg/0.1 ml with varying intervals as often as weekly for 4 weeks. Complications include cataracts, elevated intraocular pressure, granulomatous anterior uveitis, vitreous hemorrhage, and rhegmatogenous retinal detachment.
Orbital Radiation
Radiation therapy (RT) has been a common treatment modality for patients with PVRL, with a dose ranging between 30 Gy to 50 Gy in fractions of 1.5 to 2.0 Gy. In a multi-institutional study in Japan evaluating 15 patients with PIOL, all patients received RT with a median of 41 Gy, with ten receiving chemotherapy, of which three received high dose methotrexate, and nine received prophylactic cranial irradiation (PCI). Of these 15 patients, 13 had a complete remission, and the 2-year OS and disease-free survival (DFS) was 74% and 58%, respectively. Of note, PCI did not prevent CNS recurrence.
In another retrospective study of 12 patients and 21 eyes (9 with bilateral disease and 5 with CNS disease), Berenbom et al. demonstrated no recurrence in the six patients who received radiation with chemotherapy, or the one treated with radiation alone. Four patients received chemotherapy alone and two of those had ocular relapse. Milgrom et al. highlighted a lower rate of disease recurrence when systemic therapy was combined with bilateral orbital radiation. In this retrospective study, 8 patients with PVRL limited to the eye were treated with systemic chemotherapy with rituximab, high dose MTX (HDMTX), procarbazine, and vincristine followed by bilateral radiation with a median dose of 36 Gy and 2 cycles of cytarabine.
Retinopathy was reported in only two patients. A patient with advanced type 2 diabetes and secondary ocular involvement developed severe, proliferative retinopathy following ocular radiation. Another patient developed relapsed disease that infiltrated the retina. The remaining patients did not develop vision-compromising adverse effects, leading the authors to conclude that complications after orbital XRT are uncommon and the risk of toxicity may be augmented by comorbidities. Furthermore, Parsons et al. found a lower risk of radiation retinopathy when radiation is administered at doses under 45 Gy. Cataracts are a well-known complication of vitreoretinal and orbital radiation. However, cataracts are highly treatable. In addition, Milgrom et al. noted that while all patients had progressively developed cataracts, surgical extraction successfully restored their visual acuity. All of these studies are limited by small sample sizes, but in the absence of comorbidities, orbital XRT should not be withheld due to fear of vision-threatening toxicity.
Summary of Local Therapies
Due to a heterogeneous treatment schedule, lack of homogenous endpoints in the limited retrospective and prospective data, heterogeneous patient population with both secondary and primary CNS/orbital disease, a clear treatment regimen is a controversial topic. Ocular toxicities can occur with either injections or radiation resulting in long-term toxicities, but local treatment responses are favorable. While local therapies do not appear to reduce the risk of CNS dissemination, they afford excellent local control and should be utilized when ocular involvement of lymphoma is diagnosed. Decisions on which intraocular therapy should be used frontline is controversial but should involve a multidisciplinary discussion at a specialty center with experience in managing this disease. Given the rarity of this disease, prospective studies to definitively determine the optimal treatment approach for localized therapy is challenging.
Systemic Therapy
Systemic therapy may have a limited role in newly diagnosed, localized PVRL, but is frequently considered in binocular, relapsed, or in cases of CNS involvement (Figure 1). It consists of two phases, induction and consolidation. During induction and if the patient has an excellent performance status, the mainstay of therapy is a high dose MTX (HDMTX) based regimen. For consolidation, patients may receive additional chemotherapy, whole brain radiation therapy (WBRT), auto-HCT, or other therapies such as Bruton tyrosine kinase (BTKI) inhibitors.
Based on the results of the pivotal IELSG 32 (International Extranodal Lymphoma Study Group), the standard of care for induction in adults <70 years of age has shifted toward utilizing the MATRix regimen (HDMTX, cytarabine, thiotepa, rituximab). After 7 years of follow up, OS was 56% for MATRix, 37% for HDMTX, cytarabine, rituximab, and 21% for HDMTX and cytarabine. Consolidation with WBRT and autoHCT had comparable outcomes, but there was less neurocognitive dysfunction in the autoHCT group. Importantly, there were only 7 patients in this study with ocular involvement, but only one received MATRix. In patients with poorer performance status, or who cannot tolerate MATRix, then a HDMTX based regimen, with or without cytarabine and rituximab is favored.
While the benefit of systemic therapy in PCNSL is clear, whether cases of PVRL without CNS dissemination derive benefit is not clear. In a retrospective study of 59 patients, HDMTX was used in patients with PVRL with or without systemic therapy. In this cohort, only 8 also received concomitant local therapies. Complete response was achieved in 67.6%, but after a median follow up of 61 months, 71% of patients relapsed, including 29 isolated ocular relapses as the first relapse and a total of 22 CNS relapses. A recent study providing a comprehensive literature review reported comparable OS between local vs systemic therapy for PVRL without CNS disease. Interestingly data suggested that when accounting for lead time bias, OS was poorer with combination therapies. Conversely, given that 60–90% of patients will eventually develop CNS dissemination, one small prospective study of 11 patients with PVRL concluded that the combination of intravitreal MTX, HDMTX, and WBRT may reduce CNS dissemination, with only one patient at 4 years reporting CNS recurrence.
In a retrospective study by Grimm et al. of 83 patients with PIOL, there was no significant difference in relapse pattern, median progression-free survival (PFS) or OS between local treatment (MTX, ocular radiotherapy) compared to non-local therapies (systemic chemotherapy, whole brain radiotherapy). Another pooled analysis from 17 centers showed utilization of local therapy and systemic therapy was associated with no reduction in CNS recurrence and higher rates of toxicities in comparison with local therapies alone. Therefore it is controversial whether systemic therapy provided benefit in this patient population to reduce CNS dissemination.
While MATRix or other systemic regimens may be beneficial for selected patients, many reported regimens are effective in patients with relapsed disease. Given the rarity of the disease and thus limited prospective studies, the treatment landscape of PVRL remains an evolving area of research.
Autologous Hematopoietic Stem Cell Transplantation
There is limited data on the use of high dose chemotherapy and autologous stem cell transplantation (autoHCT) in the treatment of PVRL. While autoHCT is typically reserved for relapsed or refractory (R/R) PCNSL, poor general conditions of the patients as well as poor response to salvage chemotherapy may preclude consolidation therapy with autoHCT. In PCNSL, penetration of the blood-brain-barrier by myeloablative chemotherapy is important prior to autoHCT. Conditioning regimens with thiotepa have demonstrated superior outcomes compared to other protocols such as carmustine, etoposide, AraC, and melphalan (BEAM). Per the IELSG32 study, Ferreri et al. reported a similar efficacy between WBRT and autoHCT as consolidation. therapy in PCNSL with similar overall PFS at 2 years. Notably, patients who underwent WBRT experienced more neurological impairment, while patients who underwent autoHCT experienced improvement in executive function and quality of life. PRECIS demonstrated superior cognitive health after autoHCT compared to consolidation with WBRT in patients under 60 years of age with PCNSL. Furthermore, autoHCT carried an event-free survival (EFS) of 67% with autoHCT compared to 39% with WBRT at 8 years. The rate of relapse was significantly reduced with autoHCT with a hazard ratio of 0.13.
The conditioning regimen in both IELSG32 and PRECIS used a thiotepa-based protocol. The results of PRECIS suggest fit, younger patients with R/R PCNSL should be considered for consolidation with autoHCT after HDMTX-based induction therapy and autoHCT should be prioritized over WBRT given risk of neurological toxicity. Wullenkord et al. found improved PFS and OS when autoHCT was used as first-line therapy instead of a second- or third-line consolidation therapy. The retrospective analysis consisted of 247 patients with B-cell lymphoma including 45 patients with newly diagnosed PCNSL of which 10 had R/R disease. All patients received conditioning with a thiotepa-based conditioning regimen.
Chimeric Antigen Receptor T-cell Therapy
Chimeric antigen receptor (CAR-T) T cell therapy is an emerging immunomodulatory tool in cancer therapy. CAR-T therapy is often employed in relapsed or refractory large B-cell lymphoma (LBCL) and demonstrated improved PFS and OS rates compared to traditional chemoimmunotherapy regimens alone (ZUMA-1, ZUMA-7). The eyes represent an immune-privileged site and whether CAR-T cells infiltrate this site is uncertain.
Recent studies, however, have found CAR-T cells isolated in cerebrospinal fluid in patients with hematological malignancies, suggesting a route for CAR-T entry into the CNS independent of tumor antigen recognition. These studies raise important questions regarding the potential efficacy of CAR-T cell therapy in the treatment of PVRL.
Patients with B-cell malignancies with CNS involvement have been historically excluded from clinical trials with CAR-T due to concern for an elevated risk of neurotoxicity. However, there has been a renewed interest in recent years regarding the utilization of CAR-T in patients with CNS involvement of their lymphoma. In 2017, tisagenlecleucel was approved with LBCL with secondary CNS disease. Abramson et al. enrolled a 68-year-old woman with R/R DLBCL with secondary CNS lymphoma into the TRANSCEND-NHL-001 clinical trial with lisocabtagene maraleucel. The patient demonstrated recession of CNS disease and sustained remission at 12 months following infusion. No neurotoxicity was observed.
Frigault et al. treated 28 patients with secondary CNS lymphoma with CD19-directed CAR-T therapy. At 28 days, complete response was observed in 2 patients, partial response in 2 patients, and disease progression in 1 patient. At 90 days of follow-up, 3 of the 4 patients who exhibited positive response demonstrated disease control. Siddiqi et al. evaluated 5 patients with primary CNS lymphoma treated with a CD19-directed CAR-T. The median number of prior therapies was five. At 28 days after infusion, 3 out of 5 patients had complete response and 2 had stable disease. Of the 3 patients who had CR, 1 patient relapsed after 9 months of remission, 1 patient started maintenance therapy at day 43, and 1 patient continued to be followed off maintenance therapy. All patients developed greater than grade 1 cytokine release syndrome (CRS) with 2 patients requiring tocilizumab and dexamethasone. 1 patient developed grade 3 neurotoxicity; the effects were tolerable and treated supportively. Furthermore, CSF analysis revealed the CAR-T cells had successfully localized to the brain after peripheral infusion.
Several other studies have highlighted the presence of CAR-T cells in the CSF of patients treated with CD-19 CAR-T cells even in the absence of CNS-specific disease. The small cohort studies of CD19-directed CAR-T therapy in PCNSL suggest an evolving role for cellular therapy. Current clinical trials are underway to assess CD19-directed CAR therapy in primary and secondary CNS lymphoma. Future studies should include PVRL patients as a consideration.
Bruton Tyrosine Kinase Inhibitor Therapy
The elevated frequency of the IGHV4-34 gene suggests a role for antigenic stimulation of PVRL. The activated B-cell (ABC) phenotype seen in PVRL also frequently harbors mutations in CD79B and MYD88. Inhibitors of the B-cell receptor (BCR) signaling pathway may thus mitigate lymphoma cell expansion in PVRL. In a phase 2 prospective study, Guan et al. showed BTKi monotherapy demonstrated considerable success in establishing disease control after 1 month of treatment in patients with relapsed or newly diagnosed PVRL. In one month, 9 out of 10 patients demonstrated disease control and 70% achieved complete response. The median PFS was 8.3 months with no significant adverse events reported or discontinuation of therapy.
Wang et al. evaluated 3 patients with relapsed PCNSL localized to the retina without additional CNS involvement with Zanubrutinib at 160 mg twice daily for 1 month. All 3 patients demonstrated significant response with IL-10 levels returning to normal level. The authors of this study concluded Zanubrutinib may be considered as a treatment tool for relapsed PCNSL localized to the intraocular compartment to mitigate toxicities of re-exposing patients to systemic chemotherapy. All 3 patients remained in complete remission for over 6 months.
Soussain et al. conducted a phase II multicenter prospective study on Ibrutinib at 560 mg daily in patients with relapsed/refractory (R/R) PCNSL or VRL until disease progression or toxicity was observed. 44 patients were evaluated after 2 months of treatment. Disease control was observed in 70% of patients with a 19% complete response and 33% partial response. Median PFS and OS were 4.8 and 19.2 months respectively. Furthermore, Ibrutinib was detected in the cerebrospinal fluid and a positive response did not appear to be contingent on specific mutations in the BCR pathway. BTKi therapy serves as an important tool that may shift the treatment paradigm of PVRL.
Prognosis
The presence of CNS involvement is one of the most important prognostic factors in PVRL. In a meta-analysis, Zhao et al. showed a pooled 2-year survival rate of patients with PVRL with and without CNS involvement was 77% and 98% respectively. Recent studies are delving into the prognostic value of tumor cell markers in the treatment of PVRL. These markers act as surrogates of disease activity and aid in guiding treatment and overall prognosis. Chan et al. described a positive correlation between the level of IL-10 in the vitreous and the burden of malignant cells. IL-10 is an important immunomodulatory cytokine secreted by malignant B-cells. Zhao et al. found an elevated IL-10 ratio in the vitreous is seen in patients with PVRL with poor prognosis. This can serve a prognostic role in risk stratification of patients, help determine the need for more aggressive treatment, and guide therapy decisions. Prospective studies investigating whether using IL-10 levels as a surrogate for disease activity and guiding treatment can help delay or prevent CNS progression would help answer important questions in the treatment landscape of PVRL.
Conclusion
PVRL is a rare subtype of ocular lymphoma that poses significant diagnostic and treatment challenges. While diagnosis remains challenging, vitreal biopsy remains the gold standard of diagnosis, and other monitoring modalities such as OCT, MRI, and ultrasound can improve clinical response time to subtle ocular changes and disease progression. More studies are needed to determine the role of cytokine receptors in pathogenesis of PVRL but hopefully will soon provide new drug targets.
Currently, local injections of methotrexate and/or rituximab, low dose orbital radiation, and systemic chemotherapy are utilized for therapy and provide excellent 5-year survival of 98% for those without CNS involvement. Unfortunately, disease relapse or CNS dissemination occurs in over 50% of patients. For those with relapse or CNS disease, 5-year survival drops to approximately 55%, and is treated with multiagent chemoimmunotherapy regimens. Consolidation therapy includes autoHCT, WBRT, and BTKi therapy depending on patients’ access to tertiary or quaternary health care centers. CAR-T utilization in PVRL needs to be further explored.
Conflicts of Interest Statement
The authors have no conflicts of interest to declare.
Funding Statement
The authors did not receive funding.
Acknowledgements
The authors would like to thank their colleagues for their contributions to this work.
References
1. Salomão DR, Pulido JS, Johnston PB, Canal-Fontcuberta I, Feldman AL. Vitreoretinal Presentation of Secondary Large B-Cell Lymphoma in Patients With Systemic Lymphoma. JAMA Ophthalmol. 2013;131(9):1151.
doi:10.1001/jamaophthalmol.2013.334
2. Wang Y, Cheung DS, Chan CC. Case 01-2017—Primary vitreoretinal lymphoma (PVRL): report of a case and update of literature from 1942 to 2016. Ann Eye Sci. 2018;2:32-32. doi:10.21037/aes.2017.06.06
3. Kalogeropoulos D, Vartholomatos G, Mitra A, et al. Primary vitreoretinal lymphoma. Saudi Journal of Ophthalmology. 2019;33(1):66-80. doi:10.1016/j.sjopt.2018.12.008
4. Zhou M, Xu G. Recent progress in the diagnosis and treatment of primary vitreoretinal lymphoma. Taiwan J Ophthalmol. 2016;6(4):170-176. doi:10.1016/j.tjo.2016.05.002
5. Lam M, Touitou V, Choquet S, et al. Intravenous high‐dose methotrexate based systemic therapy in the treatment of isolated primary vitreoretinal lymphoma: An LOC network study. Am J Hematol. 2021;96(7):823-833. doi:10.1002/ajh.26199
6. Grimm SA, Pulido JS, Jahnke K, et al. Primary intraocular lymphoma: an International Primary Central Nervous System Lymphoma Collaborative Group Report. Annals of Oncology. 2007;18(11): 1851-1855. doi:10.1093/annonc/mdm340
7. Stacey AW, Pulido JS. The Concept of Minimal Residual Disease in the Treatment and Staging of Vitreoretinal Lymphoma. Retina. 2020; 40(7):1213-1214. doi:10.1097/IAE.0000000000002851
8. Venkatesh R, Bavaharan B, Mahendradas P, Yadav NK. Primary vitreoretinal lymphoma: prevalence, impact, and management challenges. Clin Ophthalmol. 2019;13:353-364. doi:10.2147/OPTH.S159014
9. Tang LJ, Gu CL, Zhang P. Intraocular lymphoma. Int J Ophthalmol. 2017;10(8):1301-
1307. doi:10.18240/ijo.2017.08.19
10. Coupland SE, Damato B. Understanding intraocular lymphomas. Clin Exp Ophthalmol. 2008; 36(6):564-578.
doi:10.1111/j.1442-9071.2008.01843.x
11. Coupland SE, Chan CC, Smith J. Pathophysiology of Retinal Lymphoma. Ocul Immunol Inflamm. 2009;17(4):227-237. doi:10.1080/09273940903168696
12. Di Rocco A, Petrucci L, Assanto GM, Martelli M, Pulsoni A. Extranodal Marginal Zone Lymphoma: Pathogenesis, Diagnosis and Treatment. Cancers (Basel). 2022;14(7):1742. doi:10.3390/cancers14071742
13. Soussain C, Malaise D, Cassoux N. Primary vitreoretinal lymphoma: a diagnostic and management challenge. Blood. 2021;138(17):1519-1534. doi:10.1182/blood.2020008235
14. Chan CC, Rubenstein JL, Coupland SE, et al. Primary Vitreoretinal Lymphoma: A Report from an International Primary Central Nervous System Lymphoma Collaborative Group Symposium. Oncologist. 2011;16(11):1589-1599. doi:10.1634/theoncologist.2011-0210
15. Sagoo MS, Mehta H, Swampillai AJ, et al. Primary intraocular lymphoma. Surv Ophthalmol. 2014;59(5):503-516. doi:10.1016/j.survophthal.2013.12.001
16. Fend F, Ferreri AJM, Coupland SE. How we diagnose and treat vitreoretinal lymphoma. Br J Haematol. 2016;173(5):680-692. doi:10.1111/bjh.14025
17. Hassan M, Halim MS, Afridi R, Nguyen N V., Nguyen QD, Sepah YJ. Evaluating optical coherence tomography (OCT) findings as potential biomarkers in central nervous system (CNS) lymphoma with or without ocular involvement. Int J Retina Vitreous. 2021;7(1):70. doi:10.1186/s40942-021-00345-1
18. Hochberg FH, Miller DC. Primary central nervous system lymphoma. J Neurosurg. 1988; 68(6):835-853. doi:10.3171/jns.1988.68.6.0835
19. Ahmed AH, Foster CS, Shields CL. Association of Disease Location and Treatment With Survival in Diffuse Large B-Cell Lymphoma of the Eye and Ocular Adnexal Region. JAMA Ophthalmol. 2017;135(10):1062.
doi:10.1001/jamaophthalmol.2017.3286
20. Ostrom QT, Price M, Neff C, et al. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2015–2019. Neuro Oncol. 2022;24 (Supplement_5):v1-v95. doi:10.1093/neuonc/noac202
21. Levasseur SD, Wittenberg LA, White VA. Vitreoretinal Lymphoma. JAMA Ophthalmol. 2013; 131(1):50. doi:10.1001/jamaophthalmol.2013.569
22. Coupland SE, Bechrakis NE, Anastassiou G, et al. Evaluation of vitrectomy specimens and chorioretinal biopsies in the diagnosis of primary intraocular lymphoma in patients with Masquerade syndrome. Graefe’s Archive for Clinical and Experimental Ophthalmology. 2003;241(10):860-870. doi:10.1007/s00417-003-0749-y
23. Freeman LN, Schachat AP, Knox DL, Michels RG, Green WR. Clinical Features, Laboratory Investigations, and Survival in Ocular Reticulum Cell Sarcoma. Ophthalmology. 1987;94(12):1631-1639. doi:10.1016/S0161-6420(87)33256-7
24. Falkenhagen KM, Braziel RM, Fraunfelder FW, Smith JR. B-Cells in Ocular Adnexal Lymphoproliferative Lesions Express B-cell attracting Chemokine 1 (CXCL13). Am J Ophthalmol. 2005; 140(2):335-337. doi:10.1016/j.ajo.2005.02.026
25. Shannon-Lowe C, Rickinson AB, Bell AI. Epstein–Barr virus-associated lymphomas. Philosophical Transactions of the Royal Society B: Biological Sciences. 2017;372(1732):20160271. doi:10.1098/rstb.2016.0271
26. Coupland SE. Molecular pathology of lymphoma. Eye. 2013;27(2):180-189. doi:10.1038/eye.2012.247
27. Montesinos-Rongen M, Brunn A, Bentink S, et al. Gene expression profiling suggests primary central nervous system lymphomas to be derived from a late germinal center B cell. Leukemia. 2008; 22(2):400-405. doi:10.1038/sj.leu.2405019
28. Deckert M, Brunn A, Montesinos‐Rongen M, Terreni MR, Ponzoni M. Primary lymphoma of the central nervous system—a diagnostic challenge. Hematol Oncol. 2014;32(2):57-67. doi:10.1002/hon.2087
29. Belhouachi N, Xochelli A, Boudjoghra M, et al. Primary vitreoretinal lymphomas display a remarkably restricted immunoglobulin gene repertoire. Blood Adv. 2020;4(7):1357-1366.
doi:10.1182/bloodadvances.2019000980
30. Montesinos-Rongen M, Purschke FG, Brunn A, et al. Primary Central Nervous System (CNS) Lymphoma B Cell Receptors Recognize CNS Proteins. The Journal of Immunology. 2015;195 (3):1312-1319. doi:10.4049/jimmunol.1402341
31. Touitou V, Daussy C, Bodaghi B, et al. Impaired Th1/Tc1 Cytokine Production of Tumor-Infiltrating Lymphocytes in a Model of Primary Intraocular B-Cell Lymphoma. Investigative Opthalmology & Visual Science. 2007;48(7):3223. doi:10.1167/iovs.07-0008
32. Gerstner ER, Batchelor TT. Primary Central Nervous System Lymphoma. Arch Neurol. 2010; 67(3). doi:10.1001/archneurol.2010.3
33. Hoang-Xuan K, Bessell E, Bromberg J, et al. Diagnosis and treatment of primary CNS lymphoma in immunocompetent patients: guidelines from the European Association for Neuro-Oncology. Lancet Oncol. 2015;16(7):e322-e332. doi:10.1016/S1470-2045(15)00076-5
34. Ferreri AJM, Blay JY, Reni M, et al. Relevance of intraocular involvement in the management of primary central nervous system lymphomas. Annals of Oncology. 2002;13(4):531-538. doi:10.1093/annonc/mdf080
35. Silverman RF, Abramson DH, Canestraro J, Grommes C, Francis JH. Vitreoretinal lymphoma: the importance of cerebral spinal fluid evaluation at initial diagnosis. British Journal of Ophthalmology. Published online October 8, 2024:bjo-2024-325999. doi:10.1136/bjo-2024-325999
36. Hoffman PM, McKelvie P, Hall AJ, Stawell RJ, Santamaria JD. Intraocular lymphoma: a series of 14 patients with clinicopathological features and treatment outcomes. Eye. 2003;17(4):513-521. doi:10.1038/sj.eye.6700378
37. Houillier C, Soussain C, Ghesquières H, et al. Management and outcome of primary CNS lymphoma in the modern era. Neurology. 2020; 94(10). doi:10.1212/WNL.0000000000008900
38. Chan CC, Sen HN. Current concepts in diagnosing and managing primary vitreoretinal (intraocular) lymphoma. Discov Med. 2013;15 (81):93-100.
39. Fardeau C, Lee CPL, Merle-Béral H, et al. Retinal Fluorescein, Indocyanine Green Angiography, and Optic Coherence Tomography in Non-Hodgkin Primary Intraocular Lymphoma. Am J Ophthalmol. 2009;147(5):886-894.e1. doi:10.1016/j.ajo.2008.12.025
40. Giuffrè C, Menean M, Modorati GM, et al. Primary vitreoretinal lymphoma: recent advances and literature review. Ann Lymphoma. 2020;4:17-17. doi:10.21037/aol-20-27
41. Sen HN, Bodaghi B, Hoang P Le, Nussenblatt R. Primary Intraocular Lymphoma: Diagnosis and Differential Diagnosis. Ocul Immunol Inflamm. 2009; 17(3):133-141. doi:10.1080/09273940903108544
42. Cassoux N, Giron A, Bodaghi B, et al. IL-10 Measurement in Aqueous Humor for Screening Patients with Suspicion of Primary Intraocular Lymphoma. Investigative Opthalmology & Visual Science. 2007;48(7):3253. doi:10.1167/iovs.06-0031
43. Costopoulos M, Touitou V, Golmard JL, et al. ISOLD: A New Highly Sensitive Interleukin Score for Intraocular Lymphoma Diagnosis. Ophthalmology. 2016;123(7):1626-1628. doi:10.1016/j.ophtha.2016.01.037
44. Ferreri AJM, Illerhaus G, Doorduijn JK, et al. Primary central nervous system lymphomas: EHA–ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Annals of Oncology. 2024;35(6):491-507. doi:10.1016/j.annonc.2023.11.010
45. Habot‐Wilner Z, Frenkel S, Pe’er J. Efficacy and safety of intravitreal methotrexate for vitreo‐retinal lymphoma – 20 years of experience. Br J Haematol. 2021;194(1):92-100. doi:10.1111/bjh.17451
46. Smith J. Role of intravitreal methotrexate in the management of primary central nervous system lymphoma with ocular involvementHistorical image. Ophthalmology. 2002;109(9):1709-1716. doi:10.1016/S0161-6420(02)01125-9
47. Rishi P, Manchegowda PT, Gondhale HP, et al. Intravitreal rituximab monotherapy for management of eyes with vitreoretinal lymphoma: initial experience from India. Int Ophthalmol. 2021;41(7):2495-2504. doi:10.1007/s10792-021-01805-1
48. Larkin KL, Saboo US, Comer GM, et al. Use of intravitreal rituximab for treatment of vitreoretinal lymphoma. British Journal of Ophthalmology. 2014; 98(1):99-103. doi:10.1136/bjophthalmol-2013-304043
49. Hashida N, Ohguro N, Nishida K. Efficacy and Complications of Intravitreal Rituximab Injection for Treating Primary Vitreoretinal Lymphoma. Transl Vis Sci Technol. 2012;1(3):1. doi:10.1167/tvst.1.3.1
50. Pe’er J, Hochberg FredH, Foster CS. Clinical Review: Treatment of Vitreoretinal Lymphoma. Ocul Immunol Inflamm. 2009;17(5):299-306. doi:10.3109/09273940903370755
51. Isobe K, Ejima Y, Tokumaru S, et al. Treatment of primary intraocular lymphoma with radiation therapy: A multi-institutional survey in Japan. Leuk Lymphoma. 2006;47(9):1800-1805. doi:10.1080/10428190600632881
52. Berenbom A, Davila RM, Lin HS, Harbour JW. Treatment outcomes for primary intraocular lymphoma: implications for external beam radiotherapy. Eye. 2007;21(9):1198-1201. doi:10.1038/sj.eye.6702437
53. Milgrom SA, Cheah CY, Pinnix CC, et al. Acute and late toxicity of bilateral orbital irradiation in the management of primary intraocular lymphoma. Leuk Lymphoma. 2016;57(11):2612-2618.
doi:10.3109/10428194.2016.1166490
54. Parsons JT, Bova FJ, Fitzgerald CR, Mendenhall WM, Million RR. Radiation retinopathy after external-beam irradiation: analysis of time-dose factors. International Journal of Radiation Oncology*Biology*Physics. 1994;30(4):765-773. doi:10.1016/0360-3016(94)90347-6
55. Raval V, Binkley E, Aronow ME, Valenzuela J, Peereboom DM, Singh AD. Primary central nervous system lymphoma – ocular variant: an interdisciplinary review on management. Surv Ophthalmol. 2021;66(6):1009-1020.
doi:10.1016/j.survophthal.2021.03.004
56. Kaburaki T, Taoka K, Matsuda J, et al. Combined intravitreal methotrexate and immunochemotherapy followed by reduced‐dose whole‐brain radiotherapy for newly diagnosed B‐cell primary intraocular lymphoma. Br J Haematol. 2017;179(2):246-255. doi:10.1111/bjh.14848
57. Brevet M, Garidi R, Gruson B, Royer B, Vaida I, Damaj G. First‐line autologous stem cell transplantation in primary CNS lymphoma. Eur J Haematol. 2005;75(4):288-292. doi:10.1111/j.1600-0609.2005.00508.x
58. Scordo M, Wang TP, Ahn KW, et al. Outcomes Associated With Thiotepa-Based Conditioning in Patients With Primary Central Nervous System Lymphoma After Autologous Hematopoietic Cell Transplant. JAMA Oncol. 2021;7(7):993. doi:10.1001/jamaoncol.2021.1074
59. Ferreri AJM, Cwynarski K, Pulczynski E, et al. Whole-brain radiotherapy or autologous stem-cell transplantation as consolidation strategies after high-dose methotrexate-based chemoimmunotherapy in patients with primary CNS lymphoma: results of the second randomisation of the International Extranodal Lymphoma Study Group-32 phase 2 trial. Lancet Haematol. 2017;4(11):e510-e523. doi:10.1016/S2352-3026(17)30174-6
60. Ferreri AJM, Cwynarski K, Pulczynski E, et al. Long-term efficacy, safety and neurotolerability of MATRix regimen followed by autologous transplant in primary CNS lymphoma: 7-year results of the IELSG32 randomized trial. Leukemia. 2022;36(7): 1870-1878. doi:10.1038/s41375-022-01582-5
61. Houillier C, Taillandier L, Dureau S, et al. Radiotherapy or Autologous Stem-Cell Transplantation for Primary CNS Lymphoma in Patients 60 Years of Age and Younger: Results of the Intergroup ANOCEF-GOELAMS Randomized Phase II PRECIS Study. Journal of Clinical Oncology. 2019;37(10):823-833. doi:10.1200/JCO.18.00306
62. Wullenkord R, Berning P, Niemann AL, et al. The role of autologous stem cell transplantation (ASCT) in aggressive B-cell lymphomas: real-world data from a retrospective single-center analysis. Ann Hematol. 2021;100(11):2733-2744. doi:10.1007/s00277-021-04650-5
63. Locke FL, Ghobadi A, Jacobson CA, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019;20(1):31-42. doi:10.1016/S1470-2045(18)30864-7
64. Oluwole OO, Bishop MR, Gisselbrecht C, et al. ZUMA-7: A phase 3 randomized trial of axicabtagene ciloleucel (Axi-Cel) versus standard-of-care (SOC) therapy in patients with relapsed/ refractory diffuse large B cell lymphoma (R/R DLBCL). Journal of Clinical Oncology. 2018;36(15_suppl): TPS7585-TPS7585. doi:10.1200/JCO.2018.36.15_suppl.TPS7585
65. Abramson JS, McGree B, Noyes S, et al. Anti-CD19 CAR T Cells in CNS Diffuse Large-B-Cell Lymphoma. New England Journal of Medicine. 2017;377(8):783-784. doi:10.1056/NEJMc1704610
66. Frigault MJ, Dietrich J, Martinez-Lage M, et al. Tisagenlecleucel CAR T-cell therapy in secondary CNS lymphoma. Blood. 2019;134(11):860-866. doi:10.1182/blood.2019001694
67. Siddiqi T, Wang X, Blanchard MS, et al. CD19-directed CAR T-cell therapy for treatment of primary CNS lymphoma. Blood Adv. 2021;5(20): 4059-4063. doi:10.1182/bloodadvances.2020004106
68. Kochenderfer JN, Dudley ME, Kassim SH, et al. Chemotherapy-Refractory Diffuse Large B-Cell Lymphoma and Indolent B-Cell Malignancies Can Be Effectively Treated With Autologous T Cells Expressing an Anti-CD19 Chimeric Antigen Receptor. Journal of Clinical Oncology. 2015;33 (6):540-549. doi:10.1200/JCO.2014.56.2025
69. Bishop MR, Maziarz RT, Waller EK, et al. Tisagenlecleucel in relapsed/refractory diffuse large B-cell lymphoma patients without measurable disease at infusion. Blood Adv. 2019;3(14):2230-2236. doi:10.1182/bloodadvances.2019000151
70. Guan WX, Peng XY. Vitreoretinal Lymphoma with Intraretinal Infiltration, Simulating Retinal Necrosis. Ophthalmol Retina. 2024;8(6):571-578. doi:10.1016/j.oret.2023.11.016
71. Wang L, Guan W, Peng X. Targeting Bruton Tyrosine Kinase With Zanubrutinib for Treatment of Vitreoretinal Lymphoma: Report of 3 Cases. Front Oncol. 2021;11. doi:10.3389/fonc.2021.676792
72. Soussain C, Choquet S, Blonski M, et al. Ibrutinib monotherapy for relapse or refractory primary CNS lymphoma and primary vitreoretinal lymphoma: Final analysis of the phase II ‘proof-of-concept’ iLOC study by the Lymphoma study association (LYSA) and the French oculo-cerebral lymphoma (LOC) network. Eur J Cancer. 2019;117:121-130. doi:10.1016/j.ejca.2019.05.024
73. Zhao X yu, Cheng T tian, Meng L hui, Zhang W fei, Chen Y xin. Clinical Features, Diagnosis, Management and Prognosis of Primary Intraocular Lymphoma. Front Oncol. 2022;12. doi:10.3389/fonc.2022.808511