





Targeting Aβ42 Channels to Prevent Alzheimer’s Disease
Can Early Targeting of Amyloid-beta 42 Channels Stop Alzheimer’s Disease Before it Starts?
H. Robert Guy1
- Amyloid Research Consultants
Email: [email protected]
OPEN ACCESS
PUBLISHED: 31 May 2026
CITATION: Guy, HR., 2026. Can Early Targeting of Amyloid-beta 42 Channels Stop Alzheimer’s Disease Before it Starts? Medical Research Archives, [online] 14(5).
COPYRIGHT: © 2026 European Society of Medicine. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
ISSN 2375-1924
ABSTRACT
Methods are currently being developed to diagnose early onset of Alzheimer’s Disease (AD) before symptoms appear. The next logical step is to develop treatments to prevent farther progression of the disease; i.e., to nip AD in the bud before permeant damage occurs. Here we suggest targeting amyloid beta 42 (Aβ42) oligomers and transmembrane channels to achieve that goal. Although Aβ42 channels have not been well publicized, more than thirty years of research leaves little doubt that they exist and affect both synaptic and mitochondria membranes. Experimental evidence of these claims is review here along with highly constrained concentric β-barrel molecular models of Aβ42 assemblies and their interactions with GM1 gangliosides in both aqueous and membrane environments. These models are consistent with well-developed transmembrane channel modeling criteria, are energetically sound, and are exceptionally stable throughout molecular dynamics simulations. Data used in developing these models include electron micrographs, single channel conductance measurements in both synthetic lipid bilayers and neurons, CD measurements of secondary structure in aqueous and membrane environments, NMR studies of Aβ42 assemblies in detergents, measurements of Aβ42 toxicity, and biochemical determination of which residues of Aβ42/GM1 assemblies are accessible to proteolytic cleavage.
Keywords
Amyloid-beta 42, Alzheimer’s Disease, oligomers, transmembrane channels, GM1 gangliosides
Introduction
Typically, diseases are more treatable if detected early. Promising research is underway to develop blood biomarkers to diagnose the onset of AD in middle age before symptoms appear and often decades before large amyloid plaques and tau tangles can be detected. Increases in Aβ42 concentrations and extracellular vesicle-associated Aβ42 oligomers (EV@Aβ42) are among the biomarkers. Now, ways to prevent further progression from this initial pre-Mild Cognitive Impairment (pre-MCI) stage to MCI and AD are needed. Alterations of diet, both physical and mental exercises, improvements in hearing, and non-toxic lithium may help, but treatments aimed at specific targets also should be attempted. The principal focus of this review is to explain why non-fibril assemblies of Aβ42 in conjunction with GM1 gangliosides and possibly cholesterol should be among these targets, why they are unlikely to have disordered structures, why it is important to determine their 3-dimensional structures, and suggest how they can be targeted. My group is contributing to this effort by developing structural models of Aβ42 and Aβ42/GM1 assemblies in four environments: the aqueous phase, in Aβ42/GM1 lipoproteins, on membrane surfaces including those of EVs, and as transmembrane oligomers and channels. These models are intended to serve as hypotheses to be tested experimentally and utilized in development of treatments. Experimental results supporting and used in developing these models are reviewed.
Intrinsically Disordered vs. Intrinsically Polymorphic
Most of the AD research establishment and media classify non-fibril amyloids as “Intrinsically Disordered Proteins” (IDP). This characterization can be misleading and has discouraged research on non-fibril amyloid structures. Large amyloid fibril assemblies are stabilized primarily by hydrogen bonding between β-strands of adjacent subunits. If concentration of amyloids in an aqueous phase is too low, then these bonds cannot form and the peptides are disordered. Two factors can increase order. (1) As their concentration increases, interactions between peptides become more common, and thus more stabilizing inter-subunit H-bonds can form. The number of H-bonds per peptide increases and the number of exposed hydrophobic side-chains decreases if the β-sheets of a relatively small Aβ42 cluster roll into a cylinder; i.e., a β-barrel. (2) Interactions with hydrophobic molecules can increase the content of β-secondary structure two ways: binding to hydrophobic micelles or membranes can facilitate clustering of amyloid peptides, and hydrophobic interactions can reduce backbone H-bonding to water while increasing H-bonding between backbones of adjacent peptides. NMR and EM data we use in developing and evaluating our models were obtained either in the presence of detergents, hexane, or lipid membranes. Our latest generation of non-fibril Aβ42 assemblies include GM1 ganglioside lipids. They increase the toxicity of Aβ42 assemblies, they are required for Aβ42 oligomers to trigger Ca2+ influx, they interact with soluble Aβ42 to form soluble lipoproteins, and Aβ42/GM1 assemblies bind to outer surfaces of EVs. Also, Aβ42 assemblies appear to be more ordered than those of other relatively common Aβ variants. They have a high content of β-sheet in water and 46% β-sheet plus 19% α-helix in membranes. One would expect a much lower amount of secondary structure if they were disordered.
Structural Models of Aβ42 and Aβ42/GM1 Assemblies
β-BARREL THEORY
Proteins are easier to target if their 3-D structures are known, but that has not been done for most non-fibril Aβ42 assemblies. Our group strives to fill some of this gap by developing structural models. That is feasible in the absence of high-resolution only by evoking Occam’s razor; i.e., by assuming the structures are as simple as possible. All known channel structures composed of sequentially identical subunits have structurally identical subunits. Thus, we assume that all subunits within an Aβ42 assembly have identical conformations for our 1con models and the last third of the sequence, S3 strands, form a hydrophobic antiparallel β-barrel. One of the beauties of modeling symmetric β-barrels that have only one strand per subunit is that their highly constrained backbone structures can be predicted mathematically by well-developed β-barrel theories. Strands spiral around the axis of β-barrels in a right-handed manner. In our models of S3 barrels, the tilt angle relative to the barrel’s axis, α, is limited to two values: 36° (S/N = 1.0) or 55° (S/N = 2.0) and the diameter, D, of the barrel’s backbone can be calculated if the number of strands, N, is specified.
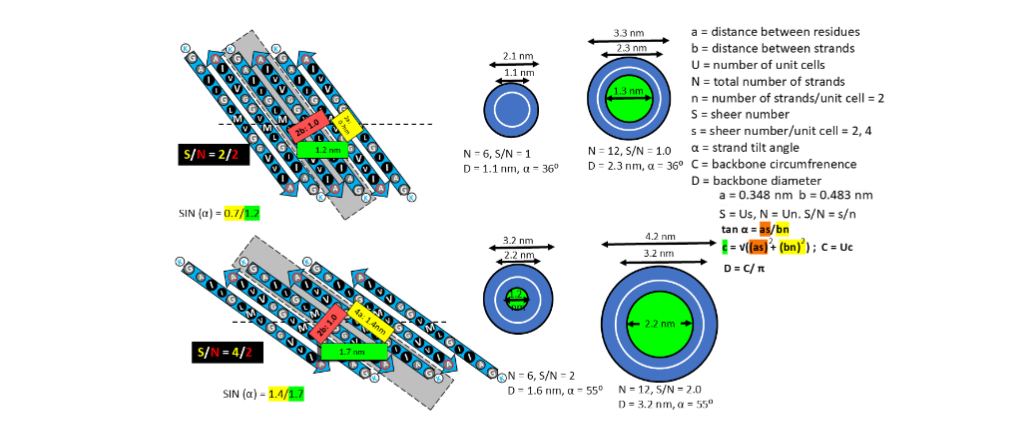
ANNULAR PROTOFIBRILS
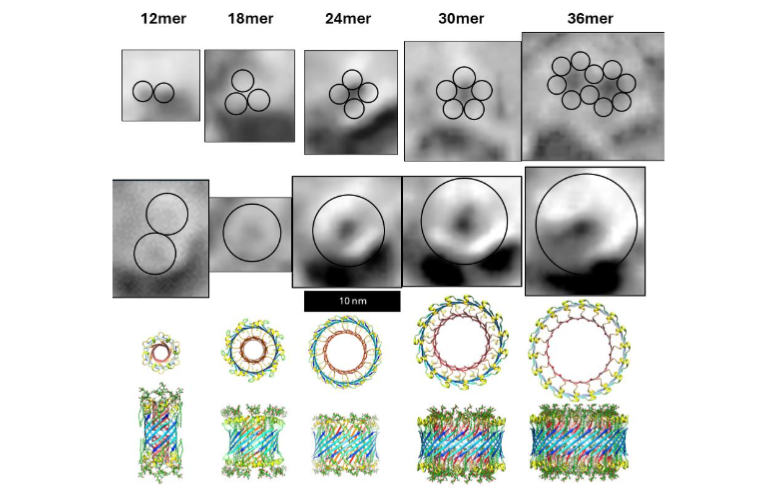
Although data suggest that non-fibril Aβ42 assemblies are highly ordered in hydrophobic environments, they also indicate that they are highly polymorphic. We first learned the extent of that polymorphism by analyzing annular protofibrils (APFs). Two types of Aβ42 APFs can be developed from soluble oligomers when hexane is present. Beaded APF (bAPFs) develop first. They resemble beads on a necklace. Our analysis of bAPF micrographs revealed five sizes of beads. We proposed that these beads correspond to soluble oligomers composed of 6, 8, 12, 16, and 18 Aβ42 subunits, and the oligomers have a concentric antiparallel β-barrel structure with S3 strands comprising core barrel and the N-terminal third, S1, and the central third, S2, of the sequence comprising an outer surrounding barrel. Hexane fills the central pore through the larger S3 barrels.
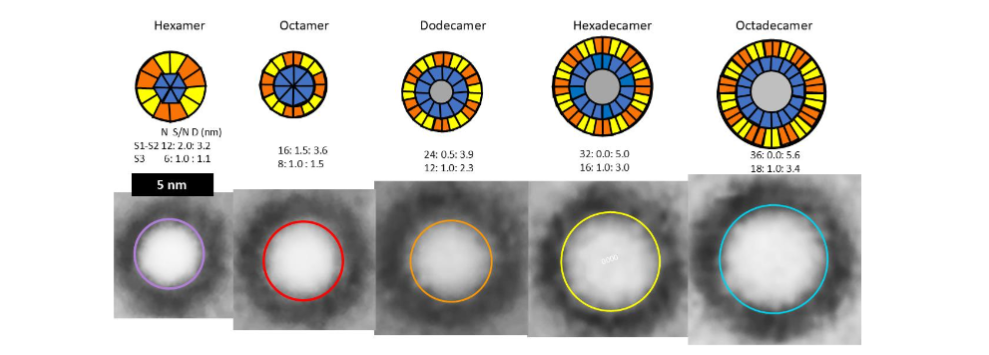
We developed atomic scale models of putative soluble oligomers with and without GM1 gangliosides. Those for the most commonly observed oligomers, putative hexamers, are illustrated in the following figure.
bAPFs morph over a period of a few days into smooth APFs (sAPFs) that have a single, often circular, wall with the same thickness independent of the highly variable sAPF diameter. We proposed that sAPFs form from bAPFs when the beads (soluble oligomers) merge to form a sAPF, which were modeled as a single set of three or four concentric β-barrels with a single hydrophobic S3 barrel sandwiched between outer barrels and inner barrels composed of S1 and S2 β-strands.
Models of Transmembrane Aβ42 Oligomers and Channels
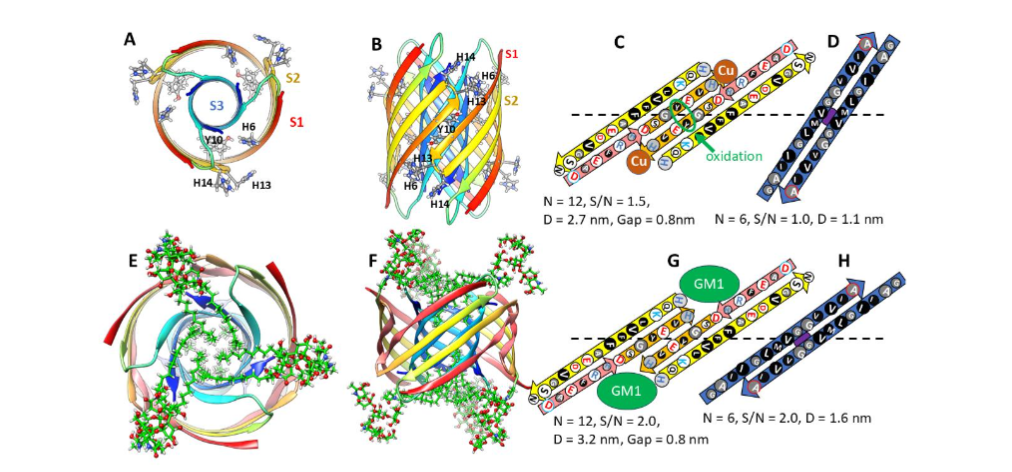
My group joined efforts to understand molecular mechanisms responsible for AD in 1994 shortly after our NIH colleges, Nelson Arispe and Harvy Pollard, discovered that Aβ42 formed channels in artificial membranes. They knew our group had modeled numerous membrane channel proteins in the absence of high-resolution data and they invited us to help them model Aβ42 channel structures. Shortly thereafter Lal’s group published atomic force microscopy of Aβ and other amyloid (α-synuclein and amylin (or islet amyloid polypeptide (IAPP)) channels. Our models have evolved since then as more data became available. We published our first Aβ42 concentric β-barrel models in 2010 and then applied this concept to α-Syn channels and Aβ42 protofibrils. The following developments since 2010 are consistent with our models and leave little doubt that Aβ42 channels exist: (1) NMR studies of Aβ42 assemblies in detergents form β-barrels with only one or two peptide conformations and S3 segments form antiparallel β-sheets with axes of P2 symmetry at position V36. (2) Cylindrin peptides with sequences of S3 fragments form 6-stranded antiparallel β-barrels. (3) Some protein toxins that exist both in the aqueous phase and as transmembrane channels have sequences similar to α-Syn and contain concentric β-barrels. (4) Relatively small Aβ42 oligomers are the only type of Aβ assembly that will form ion channels in neurons. (5) Aβ42 single channel currents have been measured in neurons from mice that have been genetically modified to create animal models of AD. (6) Aβ42 is the most toxic common Aβ42 variant. (7) Structures of Aβ42 assemblies both in water and in membranes differs substantially from those of the other most common Aβ variants, they have a higher content of regular secondary structure (β sheet in water and β + α-helix in membranes) and the side-chain of tyrosine 10 is inaccessible only in Aβ42 in either environment. (8) Aβ42 competes with VAMP2 for binding to synaptophysin of fusion pores in presynaptic membranes. (9) Aβ42 interacts with organelles including mitochondria.
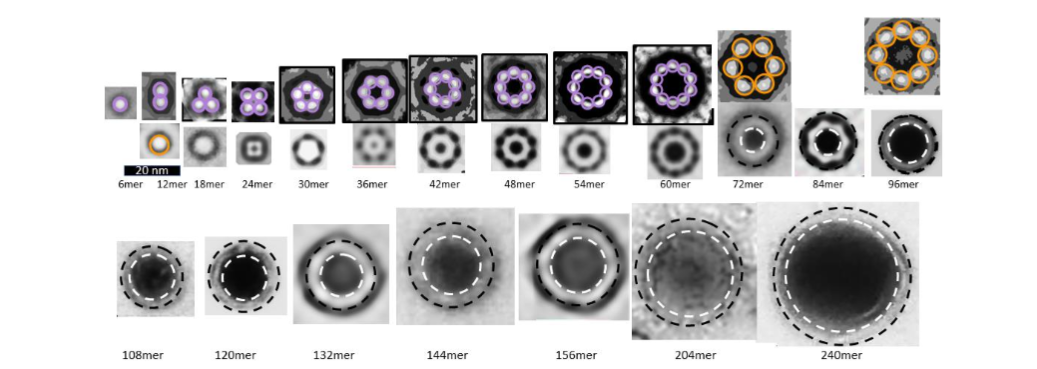
Freeze-fracture images reveal that transmembrane Aβ42 assemblies are highly polymorphic, the assemblies have many different sizes and shapes. The smallest assemblies resemble beads of bAPFs and appear in three sizes. We model these as hexamer, octamer, and dodecamer transmembrane oligomers (TMOs). Only S3 β-barrels with no water-filled pores are in the transmembrane region of TMOs. Like those of bAPFs, these beads can cluster. The freeze-fracture micrographs also include donut-shaped images of multiple sizes that we model as transmembrane channels. These resemble images of smooth APFs that develop slowly from bAPFs. As with APFs, we propose that TMOs in a cluster can merge to form channels that have only one S3 β-barrel. Our simplest models have the same basic protein folding motif regardless of the number, N, of Aβ42 subunits. S3 strands comprise an outer antiparallel β-barrel exposed to lipid alkyl chains; S2 α-helices reside on each membrane surface, and S1 β-strands or β-hairpins comprise interior β-barrels that span about half the transmembrane region from each side and that line a water-filled pore. The tilt angle, α, of S3 strands is 55° (S/N = 2.0) for dodecamers. For channels with 18 or more subunits, α can be either 36° (S/N = 1.0, Gap ~ 0.8 nm) or 55° (S/N = 2.0, Gap ~ 1.1 nm). The more tilted S3 barrel models have sufficient space between the outer S3 barrel and inner S1 barrels to contain GM1 alkyl chains. The other feature of these models that accommodates GM1 alkyl chains is that Glycine residues at every fourth position (Glycines 29, 33, and 37) fall on the same β-pleat. Since Glycine has no side-chain, this creates a groove into which GM1 alkyl chains can fit. These Glycines are conserved among all known Aβ42 sequences.
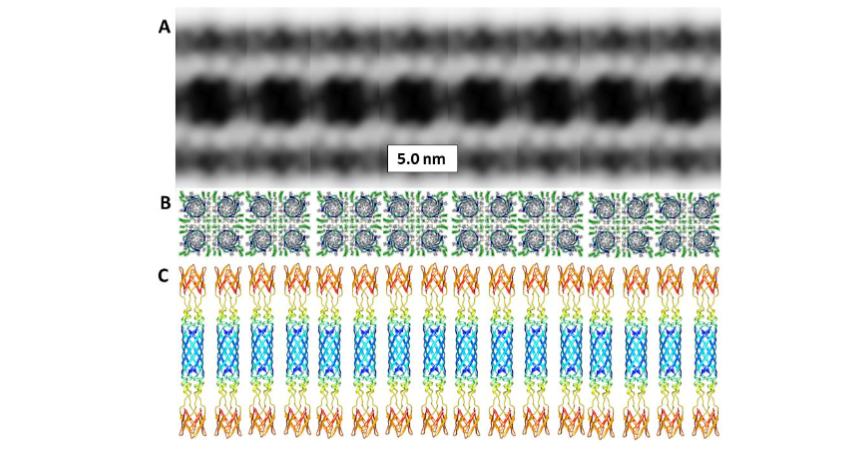
Membrane surface Aβ42/GM1 assemblies
The most compelling evidence for how Aβ42/GM1 complexes bind to the outer surfaces of membranes was obtained by Tian et al. Their cryo-electron tomography study showed that GM1 gangliosides interact with Aβ42 to form a periodic assembly that connects two liposomes. The Aβ42/GM1 assemblies do not appear to penetrate the membrane of either liposome but rather replace much of the outer leaflet of each bilayer.
Discussion
Decades of efforts to develop inexpensive, effective, and non-toxic treatments for AD that target amyloid plaques and/or tau tangles have largely failed. Most of these treatments have been attempted only after these large assemblies have been detected. But that is likely too late; the damage and degradation process may then be irreversible. One of the antibody treatments, Lecabenab, that interacts with both Aβ plaques and oligomers slows the development of AD, but does not prevent it. But it is expensive, has little effect once the treatments are stopped even though plaques are no longer present, and has toxic side-effects.
The recent development of biomarkers that identify initiation of AD before symptoms appear raise the prospect of preventing AD in its infancy before irreversible damage occurs. These biomarkers include an increase in the concentration of Aβ42 and EVs@Aβ42. Increases in the prevalence of Aβ42/GM1 lipoproteins should be added to the list of potential biomarkers. The main argument presented here is that Aβ42 oligomers and/or channels, especially those with associated GM1 gangliosides, constitute a wide range of potential targets present at this stage; e.g., drugs could be developed that target pathogenic Aβ42 assemblies or their precursors, mechanisms that remove excessive Aβ42 could be upregulated, initial formation of Aβ42 could be inhibited, or binding of GM1 gangliosides to Aβ42 could be blocked.
The situation is more complicated than most researchers realize. Structure-based drug design requires knowing the 3-D structure of the target. The first impediment is that most established AD researchers do not consider efforts to experimentally determine structures of Aβ42 channels feasible or relevant. Many do not even acknowledge they exist. Thus, structural studies of Aβ42 channels are not well funded in the US. The second impediment is polymorphism. When we began modeling Aβ42 channels we hoped to identify a relatively small number of plausible structures. The first indication that we could not do that came with our analysis of APFs. At first glance the EM images of bAPFs appeared hopelessly complicated; sizes of the beads, the number of beads per bAPF, and shapes and sizes of individual bAPFs varied greatly. Nonetheless, we were able to identify regularities among the chaos.
Aβ42 oligomers and channels are also polymorphic. Atomic force microscopy (AFM) images of Aβ42 in artificial lipid membranes detects clusters of three to six peaks around a central pore. A limitation of AFM is that it detects primarily soluble domains that extend beyond lipid headgroups. Freeze-fracture images of Aβ42 in similar bilayers reveals additional assemblies. Patch clamp recordings in neuronal membranes have identified three Aβ42 distinct single channels conductances. A biochemical analysis of Aβ42/ GM1 assemblies reported assemblies with molecular weights ranging from 45–100 kDa corresponding to assemblies of ~ 10 to 20 peptides. NMR analysis of Aβ42 in membrane-mimicking detergents identified tetramer and octamer assemblies that have two different distinct Aβ42 subunit conformations, which raises doubts about our assumption for 1Con models that all subunits have the same conformation. Another NMR study of 150 KD Aβ42 structures developed in the presence of detergent proposed that the assemblies have 32 Aβ42 subunits; but NMR analysis of these assemblies indicated that their structure closely resemble that of the tetramers. We have developed another category of models, 2con models, based on the NMR-based structure of Aβ42 tetramers. They were not included here because they are more complicated and ambiguous.
This extreme polymorphism compelled us to look for one or two relatively simply protein folding motifs that can be common to all or most polymorphs while also being consistent with most experimental data and with the channel modeling criteria we have developed over the last half century. The concentric β-barrel with both radial and P2 symmetries does that. The illustrations above show only a fraction of the structural models we have developed for more a more complete list.
Classic X-ray crystallography requires a homogeneous population of identical protein structures that can form crystals. That may prove impossible for Aβ42 channels. Fortunately, single-particle cryo electron microscopy that does not require crystals has been used to determine membrane channel structures. The third impediment is the real and perceived unreliability of protein molecular modeling done in the absence of high-resolution structural data or known structure of homologous proteins. Our group has successfully predicted gross features of numerous membrane channels and transporters, such as their secondary structures, transmembrane topology, the general position of transmembrane segments, and which segments form ion selectivity filters and toxin binding sites. However, precise details of our atomic-scale models invariably had errors when compared to subsequently determined crystal structures, making them unreliable for structure-based drug design. Our Aβ42 models of the pore could be an exception. The transmembrane portion of none of our prior channels are as constrained as our simplest Aβ42 channel models. Their potential precision is due to constraints placed on concentric β-barrels in which all subunit conformations are identical and each subunit contributes only one or two β-strands and the constraint that all concentric barrels must have the same axes of symmetry. Channels are likely to form better drug-binding sites than soluble or membrane surface assemblies because their convex pores are analogous to elongated cavities to which drugs bind. These channel models satisfy our modeling criteria exquisitely: all hydrophobic side-chains are either buried in the protein or exposed to lipid alkyl chains, no unpaired polar atoms are exposed on the outer surfaces to lipid chains, side-chains in buried regions are densely packed but do not overlap, all negatively charged carboxyl groups form salt-bridges with positively charged partners if the histidine side-chains are positively charged, most polar backbone atoms form H-bonds due to a high content of regular secondary structure that is consistent with experimental results for both channel and soluble models, the transmembrane β-barrels remain exceptionally stable throughout molecular dynamic simulations, diameters of the models are consistent with EM images, and accessibilities of segments to proteolytic cleavage are consistent with experimental data. Thus, we are optimistic that the pore regions of our models will be precise enough for structure-based drug design.
Nonetheless, researchers should, and I trust will, remain skeptical until our models are verified experimentally. Relying on the kindness of experimentalists, I have compiled a wish-list for them. It may be possible to exploit the 3-fold, 4-fold, 6-fold, 9-fold, and 12-fold radial symmetries to design channel blockers for specific channels; i.e., by developing organic ring compounds with the same symmetries that can fit snugly inside or at entrances to the pores. We suspect that larger channels are more pathogenic than smaller ones because they should be more permeable to calcium and become more prevalent as Aβ42 concentration increases. If so, larger compounds designed to bind inside the larger pores should make better drugs.
Perhaps the next most important goal is to obtain high resolution structures of Aβ42 channels. That may be easier to achieve if the polymorphism is reduced by modifying the sequence to stabilize some structures while preventing others. Our models suggest multiple ways to do that. A prime example is the Gly37Cys mutation that increases the toxicity of Aβ42 but prevents formation of fibrils by covalently linking two subunits. Our models predict that this alteration will stabilize structures in which S3 strands are highly tilted (S/N = 2) while preventing formation of channels with less tilted S3 strands. Likewise, we predict that a Val36Cys mutation will stabilize smaller diameter channels with less tilted S3 strands while inhibiting formation of channels with larger pores. It would be informative to know if these mutants form channels with different single channel conductances and whether GM1 gangliosides bind more strongly to the Gly37Cys mutant.
We have postulated two mechanisms by which gating of Aβ42 channels could be mechanosensitive. The first resembles how we proposed the mscL channels gate; stretching the membrane causes the tilts of subunits to increase, causing the taller channel’s (S/N = 1.0) pore’s diameter to increase as they transition to shorter and fatter channels for which S/N = 2.0. The second mechanism would involve the merger of smaller TMOs or channels to form larger channels. We advocate that methods used to measure gating of other mechanosensitive channels be applied to Aβ42 channels. If mechanosensitive gating occurs, will it be affected by the mutations described above to covalently link pairs of S3 strands at regions of P2 symmetry?
Segments exposed to the aqueous phase vary among the models. We have proposed stabilized peptides that mimic some exposed regions in the hope that antibodies to these peptides will bind to specific Aβ42 assemblies. If successful, some of these antibodies could be used as drugs, or at least as research tools to determine the location of specific assemblies.
The freeze-fracture images of Aβ42 assemblies in lipid bilayers that we used are far from ideal. The study, performed over 20 years ago, was terminated originally without publication because the data were deemed too complicated to be interpreted. The studies should be repeated with and without GM1 ganglioside and cholesterol present, single-particle cryo-EM should be used to obtain higher resolution images of each type of assembly, and mutated Aβ42 peptides should probably be used to reduce polymorphism. Humanin and PRP-N1 are protective against AD. We have developed, but not published, models of both interacting with Aβ42 assemblies. Experiments to determine whether either of these prevent formation of Aβ42 channels should be performed.
The report that Aβ42 competes with VAMP2 for binding to synaptophysin suggests that it is involved in the mechanism by which synaptic vesicles fuse with plasma membranes to release transmitters. Effects of Aβ42 on formation of fusion pores should be studied.
We have published concentric β-barrel models of channels composed of 18 α-Syn proteins based in part on cryo-EM and AFM images. α-Syn and Aβ42 interact, both have been proposed to contribute to transient receptor (TRP) channels of mitochondria, and both occur frequently on both sides of presynaptic plasma membranes. We have developed, but not published, hybrid channel models composed of 12 Aβ42 and 12 α-Syn peptides that resemble our α-Syn models. It should be determined whether such channels in fact occur.
Lithium has been proposed to inhibit development of AD. Our models of 1Con dodecamer channels include lithium ions bound to carboxyl groups of Asp1 and Glu3 side chains. We have proposed that Ca2+ permeation through these channels is reduced by the presence of lithium ions, and that lithium binding can be prevented by a Glu3Gln mutation. These predictions should be tested by measuring effects of lithium and the Glu3Gln mutation on single channel conductances and selectivities.
The pore is lined by S1 β-barrels in our 1Con models and by S2 β-barrels in our 2-con models. Effects of mutating charged side-chains in these segments on Aβ42 channel conductivities and selectivities should be measured to determine which category is superior.
Several mutations within the Aβ42 sequence have been discovered in patients suffering early onset AD. Effects of these mutations on Aβ42 channels should be analyzed.
Our most unorthodox proposal is that GM1 alkyl chains bind between the outer S3 barrel and pore lining barrels if the S3 strands are highly tilted; i.e., if their S/N value is 2.0. In these models the ends of the alkyl chains are near Met35 of S3. They are also near pore lining segments in the center of the transmembrane region; e.g., for 1Con models near Ala2 of 18mers and larger channels with less tilted S3 strands, and near Ser8 for channels with highly tilted S3 strands. In 2Con models the alkyl chains would end near Phe19. It may be possible to modify ends of the alkyl chains and these residues to determine if they in fact do interact.
Fantini, et. al., found that the anticancer drug Bexarptene blocks calcium-permeable Aβ42 channels. Fantini, et. al., also developed compounds with two adjacent histidines that bind to GM1 ganglioside head groups and prevent its binding to Aβ42 assemblies. They propose that these can be used as anti-AD drugs by reducing the toxicity of Aβ42 channels. Effects of their drugs on Aβ42 single channel formation and conductances and the coupling of liposomes should be tested. Can they be used in the early stage to prevent development of AD?
The initial finding by Arispi, et. al., that Aβ42 channels are permeable to calcium ions has been verified repeatedly. The calcium signaling hypothesis for AD proposes that perturbation of calcium homeostasis is a major factor in AD. Can calcium homeostasis be evaluated in pre-AD neurons? Some EVs have Aβ42 and GM1s in their outer surface. Do these EVs interact in the same way as liposomes containing Aβ42 and GM1s?
Aβ42 can interact with GM1 gangliosides and cholesterol to form soluble lipoproteins. These assemblies are consistent with the lipid-chaperone hypothesis for the interaction of Aβ42 with membranes. We know of no detailed studies of their structures. These could be informative.
This review has neglected the importance of tau and Aβ42 disposal by apolipoprotein E, microglia, and astrocytes in the pre-MCI stage. This omission should not be misconstrued as a belief that research in these areas is less important; they are simply outside my area of expertise. It would be worthwhile to determine which of the biomarkers for early development appears earliest and which most reliably predicts that AD will develop. Such studies might help determine which process or processes are initial triggers; e.g., does excessive concentration of Aβ42 trigger generation of tau?
The research I advocate will be costly. However, the expense of not preventing AD will be several orders of magnitude greater, not to mention the physical and emotional suffering of millions. This gamble is well worth making. To my colleagues and funding agencies; if you cannot or do not wish to perform these types of studies, at least support funding them.
Conflict of Interest Statement:
The author has no conflict of interests.
Funding Statement:
This work was supported in part by the Intramural Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research.
Acknowledgements:
Coordinates of our models will be made freely available at ARCGUY.org. I wish to thank Stewart R Durell and Yinon Shafrir of the National Cancer Institute for their contributions in developing the atomic scale models.
References:
- Aw TBH, Leow YJ, Kandiah N. Association of Plasma p-tau181 and Cognition in Nondemented Subjects Without Aβ42/40 Positivity. Am J Geriatr Psychiatry. 2026 Mar;34(3):386-391.
- Benussi A, Michelutti M, Lombardo TMI, Toffoletto B, Palacino F, Cenacchi V, Pelusi L, Capacchione F, Menichelli A, Perego A, Sirianni F, Cattaruzza T, Manganotti P. Diagnostic performance of plasma pTau217/Aβ42 ratio and a three-zone threshold model for Alzheimer’s disease. Neurobiol Aging. 2026 Mar;159:60-68.
- Xie P, Wang X, Wang H, Xue H, He S, Li L, Wong MS, Qiao X. Ultrasensitive extracellular vesicles-associated amyloid-β1-42 oligomers analytical platform for early diagnosis of Alzheimer’s disease. Biosens Bioelectron. 2026 Apr 15;298:118430.
- Li TR, Yao YX, Jiang XY, Dong QY, Yu XF, Wang T, Cai YN, Han Y. β-Amyloid in blood neuronal-derived extracellular vesicles is elevated in cognitively normal adults at risk of Alzheimer’s disease and predicts cerebral amyloidosis. Alzheimers Res Ther. 2022 May 12;14(1):66.
- Grant WB, Blake SM. Diet’s Role in Modifying Risk of Alzheimer’s Disease: History and Present Understanding. J Alzheimers Dis. 2023;96(4):1353-1382.
- Radić B, Blažeković A, Duraković D, Jurišić-Kvesić A, Bilić E, Borovečki F. Could Mental and Physical Exercise Alleviate Alzheimer’s Disease? Psychiatr Danub. 2021 Spring-Summer;33(Suppl 4):1267-1273.
- Li FF, Wang JP, Zhang WJ, Zhou PT, Fan M, Cai NN, Cai YF, Han K, Yang YP, Fu ZY, Yin SY, Liang BY, Han YX, Liu YH, Tong BS, Li MX, Liu YC. Trends and mechanisms of Alzheimer’s disease and hearing impairment: A 20-year perspective. Ageing Res Rev. 2025 Aug;110:102799.
- Aron L, Ngian ZK, Qiu C, Choi J, Liang M, Drake DM, Hamplova SE, Lacey EK, Roche P, Yuan M, Hazaveh SS, Lee EA, Bennett DA, Yankner BA. Lithium deficiency and the onset of Alzheimer’s disease. Nature. 2025 Sep;645(8081):712-721.
- Durell SR, Shafrir Y, Guy HR. The concentric β-barrel hypothesis for amyloids: Models of soluble and transmembrane amyloid-β42 oligomers and channels composed of identical subunits and GM1 gangliosides. bioRxiv [Preprint]. 2026 Mar 23:2026.03.19.711324.
- Korsak M, Kozyreva T. Beta Amyloid Hallmarks: From Intrinsically Disordered Proteins to Alzheimer’s Disease. Adv Exp Med Biol. 2015;870:401-21. doi: 10.1007/978-3-319-20164-1_14. PMID: 26387111.
- Cascella R, Evangelisti E, Bigi A, Becatti M, Fiorillo C, Stefani M, Chiti F, Cecchi C. Soluble Oligomers Require a Ganglioside to Trigger Neuronal Calcium Overload. J Alzheimers Dis. 2017; 60(3):923-938.
- Chakravorty A, McCalpin SD, Sahoo BR, Ramamoorthy A, Brooks CL 3rd. Free Gangliosides Can Alter Amyloid-β Aggregation. J Phys Chem Lett. 2022 Oct 13;13(40):9303-9308.
- Karkisaval, AG, Hassan, R, Nguyen, A, Balster, B, Abedin, F, Lal, R, Tatulian, SA. The structure of tyrosine-10 favors ionic conductance of Alzheimer’s disease-associated full-length amyloid-beta channels. Nat Commun. 2024; 15(1), 1296.
- Murzin, AG, Lesk, AM, & Chothia, C. Principles determining the structure of beta-sheet barrels in proteins. I. A theoretical analysis. J Mol Biol. 1994; 236(5), 1369-1381.
- Hayward, S, & Milner-White, EJ. Geometrical principles of homomeric beta-barrels and beta-helices: Application to modeling amyloid protofilaments. Proteins. 2017; 85(10), 1866-1881.
- Kayed, R, Pensalfini, A, Margol, L, Sokolov, Y, Sarsoza, F, Head, E, Hall, J, & Glabe, C. Annular Protofibrils Are a Structurally and Functionally Distinct Type of Amyloid Oligomer. Journal of Biological Chemistry. 2009; 284(7):4230-7.
- Durell, SR, Kayed, R, & Guy, HR. The amyloid concentric beta-barrel hypothesis: Models of amyloid beta 42 oligomers and annular protofibrils. Proteins. 2022; 90(5), 1190-1209.
- Williams, TL, Serpell, LC., Urbanc, B. Stabilization of native amyloid beta-protein oligomers by Copper and Hydrogen peroxide Induced Cross-linking of Unmodified Proteins (CHICUP). Biochim Biophys Acta. 2016; 1864(3), 249-259.
- Maina, MB, Al-Hilaly, YK, Serpell, LC. Dityrosine cross-linking and its potential roles in Alzheimer’s disease. Front Neurosci. 2023 17, 1132670.
- Fantini, J, Chahinian, H, Yahi, N. Progress toward Alzheimer’s disease treatment: Leveraging the Achilles’ heel of Abeta oligomers? Protein Sci. 2020; 29(8), 1748-1759.
- Arispe, N, Rojas, E, & Pollard, HB. Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: blockade by tromethamine and aluminum. Proc Natl Acad Sci U S A. (1993; 90(2), 567-571.
- Durell, SR, Guy, HR, Arispe, N, Rojas, E, & Pollard, HB. Theoretical models of the ion channel structure of amyloid beta-protein. Biophys J, 67(6). 1994; 2137-2145.
- Quist, A, Doudevski, I, Lin, H, Azimova, R, Ng, D, Frangione, B, Kagan, B, Ghiso, J, Lal, R. Amyloid ion channels: a common structural link for protein-misfolding disease. Proc Natl Acad Sci U S A. 2005; 102(30), 10427-10432.
- Shafrir, Y, Durell, SR, Arispe, N, & Guy, HR. Models of membrane-bound Alzheimer’s Abeta peptide assemblies. Proteins, 78(16). 2010; 3473-3487.
- Shafrir, Y, Durell, SR, Anishkin, A, & Guy, HR. Beta-barrel models of soluble amyloid beta oligomers and annular protofibrils. Proteins. 2010; 78(16), 3458-3472.
- Durell SR, Guy HR. The amyloid concentric β-barrel hypothesis: Models of synuclein oligomers, annular protofibrils, lipoproteins, and transmembrane channels. Proteins. 2022; 90(2):512-542.
- Serra-Batiste, M, Tolchard, J, Giusti, F, Zoonens, M, Carulla, N. Stabilization of a Membrane-Associated Amyloid-β Oligomer for Its Validation in Alzheimer’s Disease. Frontiers in Molecular Biosciences. 2018; 5, doi:10.3389.
- Ciudad, S, Puig, E, Botzanowski, T, Meigooni, M, Arango, AS, Do, J, Mayzel, M, Bayoumi, M, Chaignepain, S, Maglia, G, Cianferani, S, Orekhov, V, Tajkhorshid, E, Bardiaux, B, Carulla, N. Abeta(1-42) tetramer and octamer structures reveal edge conductivity pores as a mechanism for membrane damage. Nat Commun. 2020; 11(1), 3014.
- Muhammedkutty FNK, Prasad R, Gao Y, Sudarshan TR, Robang AS, Watzlawik JO, Rosenberry TL, Paravastu AK, Zhou HX. A common pathway for detergent-assisted oligomerization of Aβ42. Commun Biol. 2023 Nov 21;6(1):1184.
- Do, TD, LaPointe, NE, Nelson, R, Krotee, P, Hayden, EY, Ulrich, B, Quan, S, Feinstein, SC, TeFplow, DB, Eisenberg, D, Shea, JE, Bowers, MT. Amyloid beta-Protein C-Terminal Fragments: Formation of Cylindrins and beta-Barrels. J Am Chem Soc. 2016; 138(2), 549-557.
- Bode, DC, Baker, MD, & Viles, JH. Ion Channel Formation by Amyloid-β42 Oligomers but Not Amyloid-β40 in Cellular Membranes. Journal of Biological Chemistry. 2017; 292(4), 1404-1413.
- Li S, Ji X, Gao M, Huang B, Peng S, Wu J. Endogenous Amyloid-formed Ca2+-permeable Channels in Aged 3xTg AD Mice. Function (Oxf). 2023 May 26;4(4):zqad025.
- De S, Wirthensohn DC, Flagmeier P, Hughes C, Aprile FA, Ruggeri FS, Whiten DR, Emin D, Xia Z, Varela JA, Sormanni P, Kundel F, Knowles TPJ, Dobson CM, Bryant C, Vendruscolo M, Klenerman D. Different soluble aggregates of Aβ42 can give rise to cellular toxicity through different mechanisms. Nat Commun. 2019 Apr 4;10(1):1541.
- Russell CL, Semerdjieva S, Empson RM, Austen BM, Beesley PW, Alifragis P. Amyloid-β acts as a regulator of neurotransmitter release disrupting the interaction between synaptophysin and VAMP2. PLoS One. 2012;7(8):e43201.
- Swerdlow RH. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J Alzheimers Dis. 2018;62(3):1403-1416.
- Tian, Y, Liang, R, Kumar, A, Szwedziak, P, & Viles, JH. 3D-visualization of amyloid-beta oligomer interactions with lipid membranes by cryo-electron tomography. Chem Sci. 2021; 12(20), 6896-6907.
- Park, J, Simpson, C, & Patel, K. Lecanemab: A Humanized Monoclonal Antibody for the Treatment of Early Alzheimer Disease. Ann Pharmacother. 2024; 58(10), 1045-1053.
- Zhang, DY, Wang, J, Fleeman, RM, Kuhn, MK, Swulius, MT, Proctor, EA, Dokholyan, NV. Monosialotetrahexosylganglioside Promotes Early Abeta42 Oligomer Formation and Maintenance. ACS Chem Neurosci. 2022; 13(13), 1979-1991.
- Matthies D, Bae C, Toombes GE, Fox T, Bartesaghi A, Subramaniam S, Swartz KJ. Single-particle cryo-EM structure of a voltage-activated potassium channel in lipid nanodiscs. Elife. 2018; Aug 15;7:e37558.
- Bobo, C, Chaignepain, S, Henry, S, Vignaud, H, Ameadan, A, Marchal, C, Prado, E, Doutch, J, Schmitter, J M, Nardin, C, Lecomte, S, Cullin, C. Synthetic toxic Abeta(1-42) oligomers can assemble in different morphologies. Biochim Biophys Acta Gen Subj. 2017; 1861(5 Pt A), 1168-1176.
- Sukharev S, Durell SR, Guy HR. Structural models of the MscL gating mechanism. Biophys J. 2001; Aug;81(2):917-36.
- Perez-Mitta, G, & MacKinnon, R. Freestanding lipid bilayer tensiometer for the study of mechanosensitive ion channels. Proc Natl Acad Sci U S A. 2023; 120(12).
- Niikura T. Humanin and Alzheimer’s disease: The beginning of a new field. Biochim Biophys Acta Gen Subj. 2022 Jan;1866(1):130024. doi: 10.1016/j.bbagen.2021.130024. Epub 2021 Oct 7. PMID: 34626746.
- Guillot-Sestier, MV, Sunyach, C, Ferreira, ST, Marzolo, MP, Bauer, C, Thevenet, A, Checler, F. alpha-Secretase-derived fragment of cellular prion, N1, protects against monomeric and oligomeric amyloid beta (Abeta)-associated cell death. J Biol Chem. 2012;287(7), 5021-5032.
- Kim JR. Oligomerization by co-assembly of β-amyloid and α-synuclein. Front Mol Biosci. 2023 Mar 20;10:1153839.
- Amodeo GF, Pavlov EV. Amyloid β, α-synuclein and the c subunit of the ATP synthase: Can these peptides reveal an amyloidogenic pathway of the permeability transition pore? Biochim Biophys Acta Biomembr. 2021 Mar 1;1863(3):183531.
- Gao V, Briano JA, Komer LE, Burré J. Functional and Pathological Effects of α-Synuclein on Synaptic SNARE Complexes. J Mol Biol. 2023 Jan 15;435(1):167714.
- Fantini, J, Di Scala C, Yahi N, Troadec JD, Sadelli K, Chahinian H, Garmy N. Bexarotene blocks calcium-permeable ion channels formed by neurotoxic Alzheimer’s β-amyloid peptides. ACS Chem Neurosci. 2014 Mar 19;5(3):216-24.
- Wang M, Zhang H, Liang J, Huang J, Wu T, Chen N. Calcium signaling hypothesis: A non-negligible pathogenesis in Alzheimer’s disease. J Adv Res. 2025 Nov;77:513-534.
- Hu J, Linse S, Sparr E. Ganglioside Micelles Affect Amyloid β Aggregation by Coassembly. ACS Chem Neurosci. 2023 Dec 20;14(24):4335-4343.
- Tempra C, Scollo F, Pannuzzo M, Lolicato F, La Rosa C. A unifying framework for amyloid-mediated membrane damage: The lipid-chaperone hypothesis. Biochim Biophys Acta Proteins Proteom. 2022 Apr 1;1870(4):140767.
- Xia Z, Prescott EE, Urbanek A, Wareing HE, King MC, Olerinyova A, Dakin H, Leah T, Barnes KA, Matuszyk MM, Dimou E, Hidari E, Zhang YP, Lam JYL, Danial JSH, Strickland MR, Jiang H, Thornton P, Crowther DC, Ohtonen S, Bell SM, Ferraiuolo L, Mortiboys H, Higginbottom A, Wharton SB, Holtzman DM, Malm T, Ranasinghe RT, Klenerman D, De S. Co-aggregation with Apolipoprotein E modulates the function of Amyloid-β in Alzheimer’s disease. Nat Commun. 2024 Jun 1;15(1):4695.
- van Olst L, Simonton B, Edwards AJ, Forsyth AV, Boles J, Jamshidi P, Watson T, Shepard N, Krainc T, Argue BM, Zhang Z, Kuruvilla J, Camp L, Li M, Xu H, Norman JL, Vassar R, Chen J, Castellani RJ, Nicoll JA, Boche D, Gate D. Microglial mechanisms drive amyloid-β clearance in immunized patients with Alzheimer’s disease. Nat Med. 2025 May;31(5):1604-1616. doi: 10.1038/s41591-025-03574-1. Epub 2025 Mar 6. Erratum in: Nat Med. 2025 May;31(5):1712.
- Kim S, Chun H, Kim Y, Kim Y, Park U, Chu J, Bhalla M, Choi SH, Yousefian-Jazi A, Kim S, Hyeon SJ, Kim S, Kim Y, Ju YH, Lee SE, Lee H, Lee K, Oh SJ, Hwang EM, Lee J, Lee CJ, Ryu H. Astrocytic autophagy plasticity modulates Aβ clearance and cognitive function in Alzheimer’s disease. Mol Neurodegener. 2024 Jul 23;19(1):55.