





Diastolic Dysfunction in Cardiac Light-Chain Amyloidosis
Diastolic Dysfunction Unveiling Cardiac Light-Chain Amyloidosis: A Case Report
Somesh Sahai1, R. Mondal2, S. Sharma3, Biswaur Sarkar4, Julian Benito-León5,
OPEN ACCESS
PUBLISHED: 31 December 2024
CITATION: SAHA, Somesh et al. Diastolic Dysfunction Unveiling Cardiac Light-Chain Amyloidosis: A Case Report. Medical Research Archives, [S.l.], v. 12, n. 12, dec. 2024. Available at: <https://esmed.org/MRA/mra/article/view/6168>. Date accessed: 15 oct. 2025.
COPYRIGHT: © 2025 European Society of Medicine. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
DOI : https://doi.org/10.18103/mra.v12i12.6168.
ISSN 2375-1924
ABSTRACT
Background: Cardiac light-chain amyloidosis is a critical component of this multi-system disease, significantly impacting prognosis. The extent of cardiac free light-chain deposition is the primary determinant of survival.
Keywords: cardiac light-chain amyloidosis, diastolic dysfunction, prognosis
Case Presentation
The case of a 67-year-old male with a 10-year history of cardiac involvement and renal impairment is presented with a two-day history of worsening dyspnea on exertion and orthopnea. His clinical presentation was consistent with heart failure, requiring hospitalization.
Background
Cardiac amyloidosis has garnered increasing attention due to its high prevalence and significant risk of mortality in this multi-system disease. Extracellular deposition of insoluble β-sheet fibrillar proteins in the heart has been identified in 30 different types of amyloidosis. Among these, immunoglobulin-derived light chains and transthyretin are the most common culprits, causing amyloid light-chain and transthyretin cardiac amyloidosis, respectively. Amyloid light-chain amyloidosis is the more prevalent subtype.
Cardiac light-chain amyloidosis presents with symptoms such as heart failure, arrhythmias, and atrial or ventricular arrhythmias and is often misdiagnosed or diagnosed late, leading to poor outcomes.
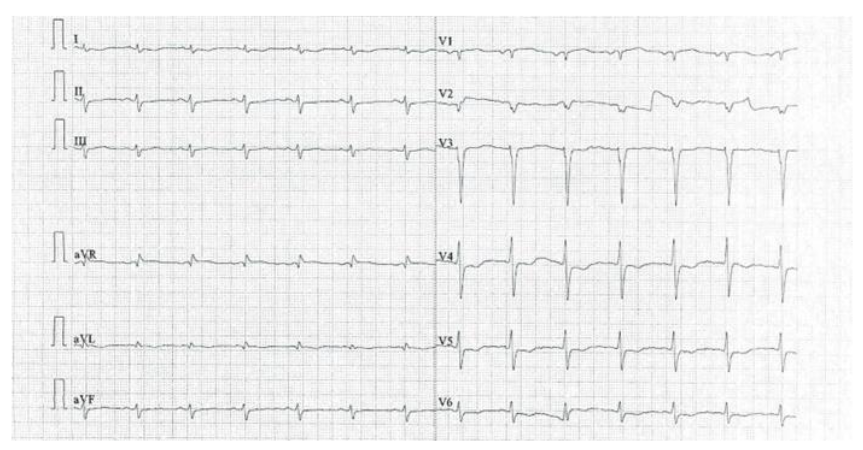
Figure 1 illustrates poor R-wave progression in V1–V4 chest leads with left ventricular hypertrophy.
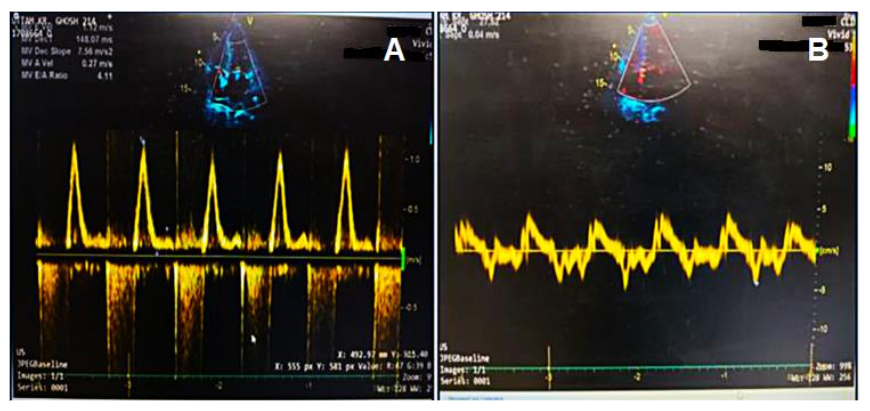
Figure 2 shows the marked increase in early diastolic mitral inflow velocity.
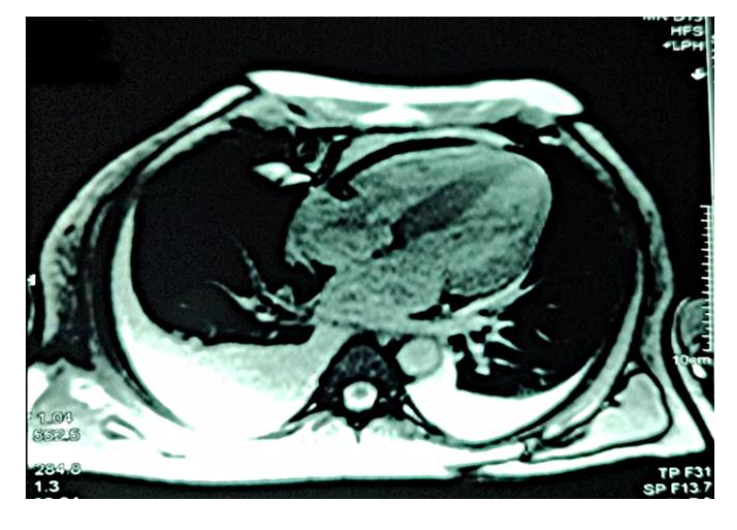
Figure 3 depicts diffuse global late gadolinium enhancement observed on cardiac magnetic resonance imaging.
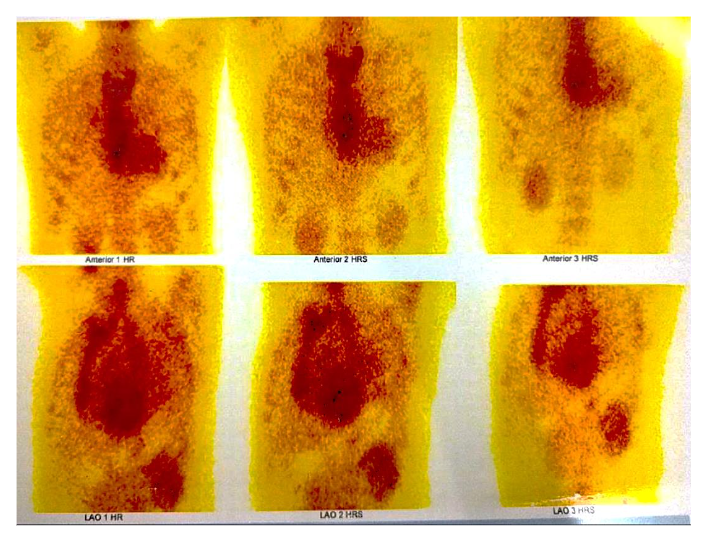
Figure 4 shows the absence of abnormal myocardial uptake in Tc-99m pyrophosphate scan.
Serum protein electrophoresis with immunofixation revealed elevated α-2, β-1, and β-2 globulins, high IgA (851 mg/dL; normal: 70–400 mg/dL), and elevated lambda free light chains (391.51 mg/L; normal: 5.71–26.30 mg/L) with a low kappa/lambda ratio (0.02). Fat pad and salivary gland biopsies were negative for amyloid deposition. Bone marrow biopsy showed 6–8% plasmacytosis without amyloid deposition. These findings supported a provisional diagnosis of free amyloid light-chain cardiac amyloidosis.
The patient was referred to a hematologist for targeted chemotherapy. Given the high suspicion of cardiac involvement and the evidence of inducible ischemia on stress echocardiography, percutaneous coronary revascularization was prioritized over autologous stem cell transplantation due to the increased risk of complications with coronary artery bypass grafting. Following revascularization, the patient initiated chemotherapy and was discharged with recommendations for regular hematological and cardiological follow-ups.
Discussion
Amyloidosis is a systemic disorder marked by extracellular deposition of insoluble β-sheet fibrillar proteins, resistant to proteolytic cleavage, causing progressive organ dysfunction. Over 30 precursor proteins are implicated in amyloid formation, often associated with proteoglycans and serum amyloid P. Among these, cardiac involvement stands as the most critical prognostic factor, predominantly driven by two protein types: immunoglobulin-derived light chains (amyloid light-chain cardiac amyloidosis) and transthyretin (transthyretin cardiac amyloidosis). Amyloid light-chain cardiac amyloidosis remains more prevalent, with an estimated incidence of 8–12 cases per million.
Historically considered rare, transthyretin cardiac amyloidosis is now being recognized more frequently, as highlighted by large autopsy series. Despite advancements in diagnostic tools, the heterogeneous presentation of cardiac amyloidosis often leads to delayed diagnoses, with dyspnea on exertion being the most common clinical symptom. Other nonspecific features, such as fatigue and low blood pressure, further complicate timely identification.
The diagnostic workup demands integration of clinical findings with advanced imaging modalities, biochemical markers, and tissue biopsies. Key electrocardiographic findings include low voltage QRS complexes and left ventricular hypertrophy, while echocardiography can reveal hallmark red flags such as restrictive filling patterns and concentric hypertrophy.
Cardiac MRI is a pivotal tool for detecting early amyloid deposition, particularly in patients with inconclusive echocardiographic findings. The capacity of cardiac MRI to identify diffuse late gadolinium enhancement provides invaluable insight into disease severity and extent of myocardial involvement. In this case, a comprehensive diagnostic approach confirmed amyloid light-chain cardiac amyloidosis through elevated lambda-free light chains, a low kappa/lambda ratio, and evidence of plasma cell dyscrasia from bone marrow biopsy. The absence of myocardial uptake on Tc-99m pyrophosphate scintigraphy excluded transthyretin cardiac amyloidosis, further solidifying the diagnosis.
Management of cardiac light-chain amyloidosis hinges on early detection and targeted treatment. For amyloid light-chain cardiac amyloidosis, chemotherapy aims to suppress the clonal plasma cell population driving amyloidogenesis. In our patient, concurrent coronary artery disease presented additional therapeutic challenges, necessitating revascularization before initiating chemotherapy. This multidisciplinary approach underscores the complexity of managing cardiac amyloidosis, particularly in cases with overlapping comorbidities.
Despite therapeutic advancements, cardiac amyloidosis remains associated with significant morbidity and mortality. Increased awareness among clinicians is essential to facilitate early diagnosis, enabling timely initiation of therapies that can stabilize disease progression and improv
Conclusion
This case highlights the diagnostic and therapeutic challenges in managing cardiac light-chain amyloidosis, particularly amyloid light-chain cardiac amyloidosis, a rare but life-threatening condition. Early detection through advanced imaging, laboratory tests, and tissue biopsies is critical for optimal management. Multidisciplinary care, integrating cardiology, hematology, and oncology expertise, is pivotal in addressing the complex needs of these patients. Continued research and heightened clinical vigilance are necessary to improve early recognition and therapeutic strategies, ultimately enhancing patient outcomes in cardiac light-chain amyloidosis.
Author’s contribution:
All authors contributed significantly to the creation of this manuscript; each fulfilled the criterion established by the ICMJE.
Conflict of Interest:
None
Funding Statement:
None.
Acknowledgements:
J. Benito-León is supported by the National Institutes of Health, Bethesda, MD, USA (NINDS #R01 NS39422), the European Commission (grant ICT-2011-287739, NeuroTREMOR), and The Recovery, Transformation and Resilience Plan at the Ministry of Science and Innovation (grant TED2021-130174B-C33, NETremor).
Ethics statement:
Written informed consent was obtained from the
References
2. Falk RH, Alexander KM, Liao R, Dorbala S. AL (Light-Chain) Cardiac Amyloidosis: A Review of Diagnosis and Therapy. J Am Coll Cardiol. 2016;68(12):1323-1341. doi:10.1016/j.jacc.2016.06.053
3. Manolis AS, Manolis AA, Manolis TA, Melita H. Cardiac amyloidosis: An underdiagnosed/ underappreciated disease. Eur J Intern Med. 2019;67:1-13. doi:10.1016/j.ejim.2019.07.022
4. Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349(6):583-596. doi:10.1056/NEJMra023144
5. Porcari A, Falco L, Lio V, et al. Cardiac amyloidosis: do not forget to look for it. Eur Heart J Suppl. 2020;22(Suppl E):E142-E147. doi:10.1093/eurheartj/suaa080
6. Mohammed SF, Mirzoyev SA, Edwards WD, et al. Left ventricular amyloid deposition in patients with heart failure and preserved ejection fraction. JACC Heart Fail. 2014;2(2):113-122. doi:10.1016/j.jchf.2013.11.004
7. Porcari A, Pagura L, Rossi M, et al. Light-chain cardiac amyloidosis: A case report of extraordinary sustained pathological response to cyclophosphamide, bortezomib, and dexamethasone combined therapy. Eur Heart J Case Rep. 2022;6(4):ytac1307. doi:10.1093/ehjcr/ytac1307
8. Merlo M, Porcari A, Pagura L, et al. A national survey on prevalence of possible echocardiographic red flags of amyloid cardiomyopathy in consecutive patients undergoing routine echocardiography: Study design and patients’ characterization—The first insight from the AC-TIVE Study. Eur J Prev Cardiol. 2022;29(5):e173-e177. doi:10.1093/eurjpc/zwab127
9. Fontana M, Chung R, Hawkins PN, Moon JC. Cardiovascular magnetic resonance for amyloidosis. Heart Fail Rev. 2015;20(2):133-144. doi:10.1007/s10741-014-9470-7
10. Cai S, Haghbayan H, Chan KKW, et al. Tissue mapping by cardiac magnetic resonance imaging for the prognostication of cardiac amyloidosis: A systematic review and meta-analysis. Int J Cardiol. 2024. doi:10.1016/j.ijcard.2024.131892. PMID: 38382853
11. Dorbala S, Cuddy S, Falk RH. How to Image Cardiac Amyloidosis: A Practical Approach. JACC Cardiovasc Imaging. 2020. doi:10.1016/j.jcmg.2019.07.015. PMID: 31607664; PMCID: PMC7148180
12. Isomali D, Mohty D, Grogan M, et al. Treatment of amyloid light chain cardiac amyloidosis: Systematic review and future directions. Clin Adv Hematol Oncol. 2022. PMID: 36206073
13. Palladini G, Milani P. Diagnosis and Treatment of AL Amyloidosis. Drugs. 2023 Feb;83(3):203-216. doi: 10.1007/s40265-022-01830-z. Epub 2023 Jan 18. PMID: 36652193.
14. Gertz MA. Immunoglobulin light chain amyloidosis: 2022 update on diagnosis, prognosis, and treatment. Am J Hematol. 2022. doi:10.1002/ajh.26569. PMID: 35429180
15. Palladini G, Milani P, Merlini G. Management of AL amyloidosis in 2020. Blood. 2020 Dec 3;136(23):2620-2627. doi: 10.1182/blood.2020006913. PMID: 33270858.
16. Donnelly JP, Hanna M. Cardiac amyloidosis: An update on diagnosis and treatment. Cleve Clin J Med. 2017 Dec;84(12 Suppl 3):12-26. doi: 10.3949/ccjm.84.s3.02. PMID: 29257735.