






Direct Reprogramming of Human Cells into Insulin Producers
Direct Epigenetic Reprogramming of Human Somatic Cells into Insulin-Producing Cell
Raza Ali Naqvi 1, Amar Singh 2, Afsar R Naqvi 1, Medha Priyadarshini 3, Sujata Prasad 4
- University of Illinois at Chicago, USA
- Department of Surgery, University of Minnesota, USA
- Department Medicine, University of Illinois at Chicago
- MLM Lab, 3510 Hopkins Place N, Minnesota, USA
OPEN ACCESS
PUBLISHED 30 September 2025
CITATION Naqvi, RA., Singh, A., et al., 2025. Direct Epigenetic Reprogramming of Human Somatic Cells into Insulin-Producing Cell. Medical Research Archives, [online] 13(9). https://doi.org/10.18103/mra.v13i9.6852
COPYRIGHT © 2025 European Society of Medicine. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
DOI https://doi.org/10.18103/mra.v13i9.6852
ISSN 2375-1924
ABSTRACT
Restoring insulin production through replacement of pancreatic β-cells presents a promising strategy for treating individuals with type 1 diabetes. However, current methods involving induced pluripotent stem cell differentiation are often time-consuming, multi-stage, and limited by safety and efficiency concerns. To overcome these challenges, we developed a simplified and direct strategy to convert human somatic cells into insulin-producing cells using an epigenetic activation system. This system combines a multiplex epigenetic engineering vector composed of dCas9.P300core and guide RNAs targeting five key β-cell genes: PDX1, NKX6.1, MAFA, Insulin, and glucose transporter type 2 (Glut2). The resulting Glut2⁺ cells exhibited glucose-responsive insulin secretion and expressed essential β-cell transcription factors including NKX2.2, along with insulin-processing and secretory machinery genes (Cav1.3, GSK3β, KCNJ11, SLC30A8). Absence of α-cell markers (aristaless-related homeobox or glucagon) confirmed lineage specificity and functional fidelity. This reprogramming approach eliminates the need for pluripotent intermediates and significantly reduces the time required to generate functional β-like cells. Our platform offers a rapid, non-integrative, and scalable method for producing insulin-secreting cells, with potential applications in personalized cell therapy, disease modeling, and high-throughput drug screening for diabetes research.
Keywords
epigenetic reprogramming, insulin-producing cells, somatic cells, type 1 diabetes, CRISPR, gene activation
INTRODUCTION
Restoration of insulin-producing pancreatic β-cells holds transformative potential for treating type 1 diabetes (T1D), a condition characterized by autoimmune destruction of native β-cells. While allogeneic islet transplantation has demonstrated clinical success in reestablishing glycemic control without exogenous insulin, this approach remains constrained by severe donor shortages and lifelong immunosuppression requirements. Xenotransplantation using porcine islets has shown promise in non-human primate models, yet safety concerns related to porcine endogenous retroviruses (PERVs) and zoonotic transmission continue to limit clinical translation. To circumvent donor limitations, efforts have turned toward generating insulin-producing cells from induced pluripotent stem cells (iPSCs). However, these methods are technically complex, involve multi-stage differentiation spanning 4–6 weeks, require precisely timed cytokine and small molecule exposure, and carry risks of teratoma formation or off-target lineage differentiation. These limitations underscore the urgent need for simpler, scalable, and safer approaches to generate β-like cells directly from accessible human somatic sources.
Recent advances in CRISPR-based epigenetic engineering have enabled direct gene activation without introducing double-strand breaks or integrating foreign DNA. In particular, the use of a catalytically inactive Cas9 (dCas9) fused to the P300core histone acetyltransferase domain has demonstrated potent transcriptional activation of silent genes by remodeling local chromatin. Unlike traditional trans activators, the P300core domain enhances acetylation of H3K27, allowing for robust transcription even in the absence of endogenous transcription factors. A landmark study demonstrated that dCas9.P300core can activate target genes—even in the absence of their native transcription factors—by guiding histone acetylation at promoter regions.
Building on this foundation, we hypothesized that targeted activation of multiple β-cell lineage regulators—pancreatic and duodenal homeobox 1 (PDX), NK6 homeobox 1 (NKX6.1), musculoaponeurotic fibrosarcoma oncogene homolog A (MAFA), Insulin, and Glut2—could convert somatic cells into glucose-responsive insulin-producing cells. We therefore engineered a Multiplex Epigenetic Engineering Vector-β (MEEV-β) that integrates all necessary guide RNAs with the dCas9.P300core fusion in a single delivery platform. This approach bypasses pluripotent intermediates, eliminates the need for staged cytokine cues, and achieves lineage-specific β-cell gene expression in just seven days. Importantly, the absence of α-cell lineages marker such as aristaless-related homeobox (ARX) or glucagon (GCG), expression supports the specificity of conversion. This proof-of-concept lays the foundation for a new class of programmable, non-integrative β-cell engineering tools with translational potential in autologous therapy, disease modeling, and drug screening for T1D.
METHODS
Cell Culture
Human somatic cells used in this study included lung endothelial cells (CD31⁺), aortic endothelial cells (CD31⁺), lung fibroblasts, and peripheral blood mononuclear cells (PBMCs). Endothelial cells were cultured in EGM-2 or EGM-2 MV medium (Lonza) on collagen-coated dishes. Lung fibroblasts were maintained in DMEM supplemented with 10% FBS, 1% Penicillin-Streptomycin, and 1% antimycotic agent. PBMCs were isolated using Ficoll-Paque Plus and stimulated with PHA (5 µg/mL) for 48 hours prior to transfection.
Cloning and assembly of the MEEV-β guide RNA design and cloning
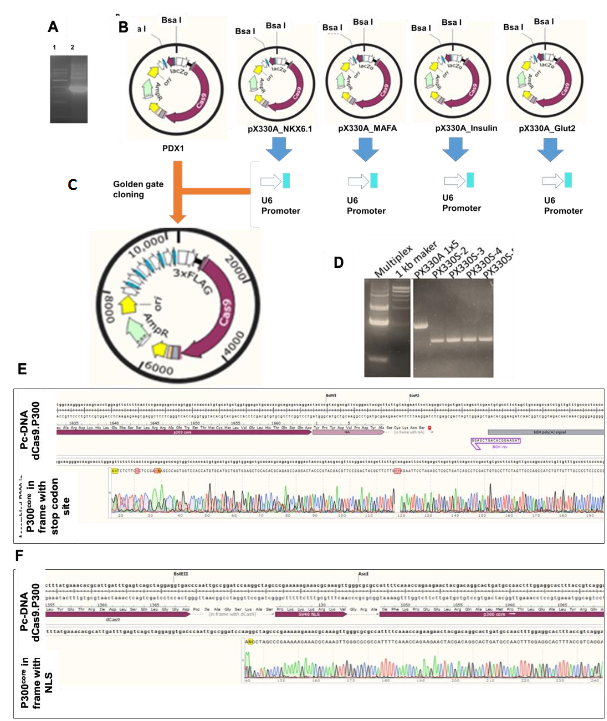
gRNAs targeting DNase I hypersensitive promoter regions of PDX1, NKX6.1, MAFA, Insulin, and Glut2 were designed using the CRISPR direct tool. The most effective gRNAs (determined via RT-qPCR) were cloned into Addgene vectors: PDX1 into pX330A-1×5, NKX6.1, MAFA, Insulin, and Glut2 into pX330S-2, -3, -4, -5. To generate a single multiplex construct, gRNA-expressing plasmids were assembled using Golden Gate cloning. Following the manufacturer’s instructions for Golden Gate assembly, the reaction mixture was prepared using: 1.5 µL each of 100 ng/µL plasmids pX330S-2 (NKX6.1), pX330S-3 (MAFA), pX330S-4 (Insulin), and pX330S-5 (Glut2); 1.5 µL of 50 ng/µL pX330A-1×6 (PDX1); 2 µL of 10× T4 DNA Ligase Buffer; 1 µL of BsaI; 1 µL of Quick Ligase; and 8.5 µL of nuclease-free water, bringing the total reaction volume to 20 µL. The mixture was subjected to thermal cycling at 37°C for 5 min and 16°C for 10 min, repeated for 25 cycles, to facilitate assembly. Assembled plasmids were transformed into TOP10 competent E. coli and verified by colony PCR and sequencing. Purified plasmid was screened for golden gate assembly by PCR.
Fusion of P300core into dCas9 vector
To enable epigenetic activation, the P300core domain was cloned in-frame into the pX330A-dCas9 1×6 backbone using HiFi DNA assembly. The NLS was removed from dCas9 by EcoRI and FseI digestion. P300core was PCR-amplified from pcDNA-dCas9.P300core. The assembled product was transformed into Stbl3 E. coli cell to prevent recombination and confirmed by sequencing. The final construct, named MEEV-β, contained dCas9.P300core and five gRNAs targeting β-cell genes.
In-frame Cloning of P300core into the Multiplex dCas9 Vector
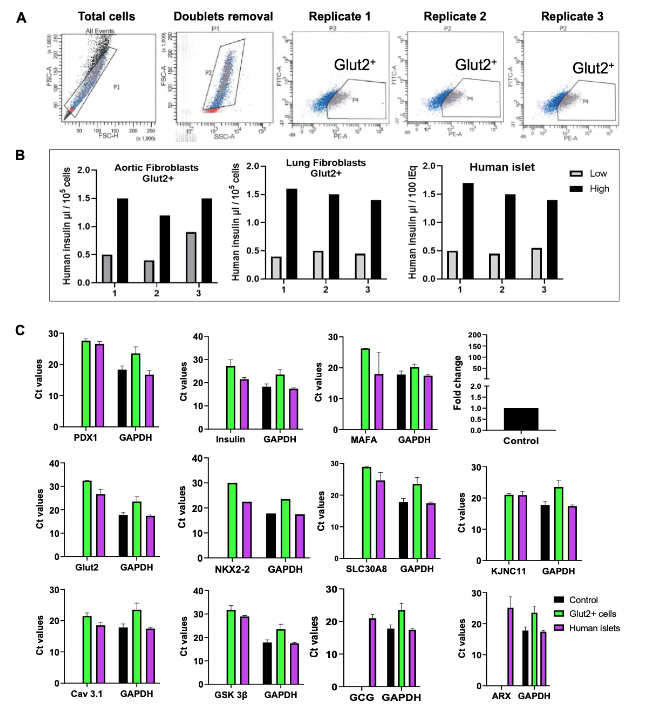
To construct the MEEV-β vector, the P300core domain was cloned in-frame into the dCas9 region of the pX330A-dCas9 1×6 plasmid containing gRNAs for PDX1, NKX6.1, MAFA, Insulin, and Glut2. First, the nuclear localization signal (NLS) was removed from the dCas9 cassette using EcoRI and FseI restriction digestion. Simultaneously, the P300core domain was PCR-amplified from the pcDNA-dCas9.P300core vector using Q5 High-Fidelity 2X Master Mix. Each 50 µL PCR reaction contained: 25 µL Q5 Master Mix, 5 µL 10 µM forward primer, 5 µL 10 µM reverse primer, 200 ng template DNA (pcDNA-dCas9.P300core) and nuclease-free water to 50 µL. The thermal cycling conditions were: Initial denaturation: 98°C for 30 sec, 35 cycles of 98°C for 10 sec, 72°C for 30 sec, final extension: 72°C for 15 min and hold at 4°C. The PCR product was purified using a gel extraction kit. For vector assembly, 50 ng of linearized vector and 100 ng of purified P300core PCR product were mixed with NEBuilder HiFi DNA assembly master mix and incubated at 50°C for 1 hour. The assembled vector was transformed into One Shot® Stbl3™ Chemically Competent E. coli to minimize recombination within the repetitive P300core sequence. Correct in-frame cloning was confirmed by PCR and sequencing using primers flanking the left and right junctions. The verified MEEV-β plasmid was then used to transfect lung endothelial cells, lung fibroblasts, aortic endothelial cells, and human PBMCs. After 14 days, Glut2⁺ cells were enriched via flow cytometry using anti-Glut2-PE.
Quantitative RT-PCR
Total RNA was extracted from transfected and flow-sorted Glut2⁺ cells using the RNeasy Plus Micro Kit. cDNA was synthesized using the iScript™ cDNA Synthesis Kit in a 20 µL reaction. Thermal cycling for cDNA synthesis: 25°C for 5 min (priming), 46°C for 20 min (reverse transcription), 90°C for 1 min (enzyme inactivation) and hold at 4°C. Real-time PCR was performed using SsoAdvanced™ Universal SYBR Green Supermix on a CFX96 Real-Time PCR System.
Glucose stimulated Insulin secretion
Flow sorted Glut2+ cells were plated in collagen coated 96 well plates (1000 cells each well). After 48h cells showed the signs of attachment and the cells were carefully washed in Krebs buffer containing 2mM glucose. Following are the components of the stock solution of Krebs buffer: 128 mM NaCl, 5 mM KCl, 2.7 mM CaCl2 dihydrate, 1.2 mM MgCl2, 1 mM Na2HPO4, 1.2 mM KH2PO4, 5 mM NaHCO3, 10 mM HEPES, and 0.1% BSA in deionized water. After washing, the cells were incubated for 2hr in 2 mM glucose prepared in Krebs buffered. The cells were then washed again and incubated sequentially under following conditions: 30 min each with 2.8 mM, 28 mM, 2 mM, 28 mM, 2 mM, and 20 mM glucose in Krebs buffer. The insulin secretion from these supernatants was detected as per instructions of Human Ultrasensitive Insulin ELISA.
RESULTS
Rational design for β-cell programming via epigenetic activation
Previous studies have established that the transcription factors PDX1, NKX6.1, and MAFA serve as master regulators of pancreatic β-cell development and function. These factors are essential for activating downstream gene networks involved in insulin biosynthesis, storage, and glucose responsiveness. To harness this biology for direct somatic cell reprogramming, we prioritized these genes as primary epigenetic targets for activation in non-β cells. Under physiological conditions, PDX1, NKX6.1, and MAFA loci remain epigenetically repressed in non-β cells, typically residing in heterochromatin regions. To enable transcriptional activation, we mapped DNase I hypersensitive sites (DHSs) near the promoters of each gene—particularly within 100 bp upstream of the transcription start site and enriched for H3K4me1 histone marks—regions known to favor chromatin remodeling. These DHS regions were selected to design multiple guide RNAs (gRNAs) targeting the transcriptional regulatory elements. Each gRNA was cloned into the pSPgRNA backbone and co-transfected with the pcDNA-dCas9.P300core vector into human somatic cells. After 4 days, qRT-PCR revealed robust transcriptional activation of the targeted genes. The top-performing gRNAs—gRNA2 (PDX1), gRNA3 (NKX6.1), and gRNA3 (MAFA)—were selected for subsequent experiments.
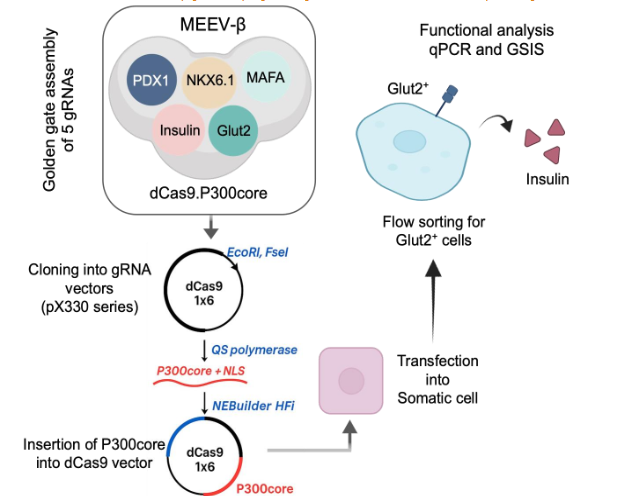
Development of Multiplex Epigenetic Engineering Vector (MEEV-β)
To efficiently co-deliver all five gRNAs and the dCas9.P300core activator in a single construct, we created a custom multiplex epigenetic engineering vector, termed MEEV-β. While multiplex CRISPR systems have been widely implemented for gene editing, their adaptation for epigenetic modulation—particularly using dCas9.P300core—had not been demonstrated.
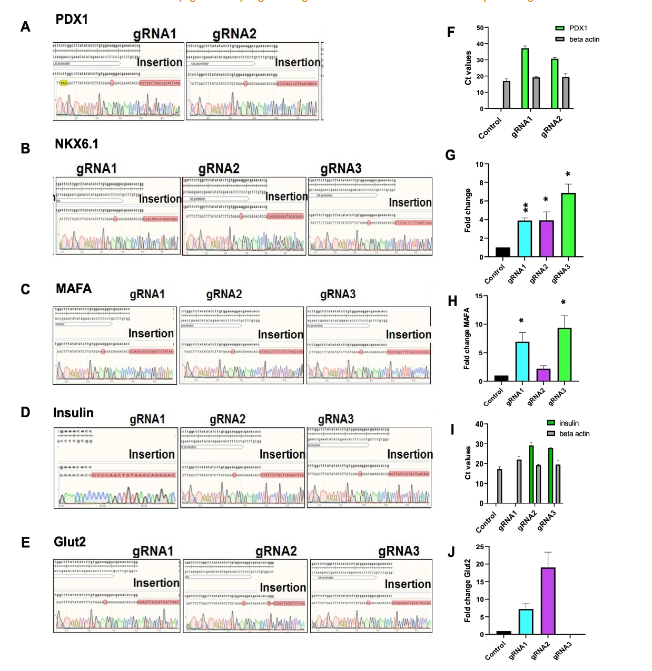
We began by modifying the pX330A-dCas9 1×5 vector backbone to accommodate in-frame insertion of the P300core domain. This involved: 1) Removing the nuclear localization signal (NLS) from dCas9 via restriction digestion. 2) PCR amplification of P300core (with an NLS) from the pcDNA-dCas9.P300core plasmid, using primers overlapping the N- and C-termini of dCas9 and the vector backbone. 3) Assembly of the P300core insert into the linearized vector using HiFi DNA assembly, confirmed by junctional sequencing. After verifying the correct in-frame fusion, we proceeded to clone the five optimal gRNAs into pX330A-dCas9.P300core (1×5) and accompanying vectors using Golden Gate assembly. The final construct—MEEV-β—thus comprised one vector delivering the dCas9.P300core fusion protein and all five gRNAs targeting PDX1, NKX6.1, MAFA, insulin, and Glut2.
Functional validation: Glucose-responsive insulin secretion
Human somatic cells from our in-house cell bank were transfected with the MEEV-β construct. After 7 days, Glut2+ cells were enriched by flow sorting, as Glut2 expression is a reliable surrogate for β-cell glucose sensitivity. Sorted cells were then challenged with low (2.8 mM) and high (28 mM) glucose concentrations in Krebs buffer to assess glucose-stimulated insulin secretion (GSIS). The Glut2+ cells exhibited a 1.5-fold increase in insulin secretion at high glucose levels compared to low glucose conditions, confirming their functional β-cell-like phenotype.
Molecular characterization of Glut2⁺ β-like cells
To further validate the β-cell identity of MEEV-β-derived Glut2⁺ cells, we performed RT-qPCR analysis of genes involved in insulin processing, vesicle trafficking, and glucose-stimulated insulin secretion. These included NKX2.2 (a downstream transcription factor of PDX1 and MAFA), as well as β-cell functional genes such as SLC30A8, KCNJ11, GSK3β, and Cav1.3. Importantly, Glut2+ cells lacked expression of α-cell markers, such as ARX and GCG, confirming lineage specificity and eliminating concerns about off-target endocrine fate. The expression profile was most consistent and robust in fibroblast-derived cells, suggesting that fibroblasts represent an optimal somatic source for β-cell reprogramming using this platform.
DISCUSSION
Conventional strategies for generating insulin-producing β-like cells typically involve multi-step differentiation protocols starting from iPSCs or human embryonic stem cells (hESCs). These methods often require 4 to 6 weeks of tightly timed culture conditions, sequential exposure to stage-specific cytokines and growth factors and specialized extracellular matrices to mimic developmental cues. While powerful, these approaches have limitations: they are labor-intensive, require multiple cytokines and developmental cues, and carry risks linked to any residual pluripotent cells, such as tumorigenicity and teratoma formation. In contrast, our MEEV-β platform introduces a streamlined, 7-day single-step conversion from primary somatic cells directly into Glut2⁺ insulin-producing cells. By targeting five essential β-cell lineage-defining genes (PDX1, NKX6.1, MAFA, Insulin, GLUT2) via a dCas9.P300core-based multiplexed epigenetic editing strategy, we eliminate the need for pluripotency induction, transgene integration, or exogenous cytokine supplementation. This dramatically reduces time, cost, and safety risks, while maintaining high efficiency of conversion. Notably, we observed a complete absence of α-cell markers such as ARX and GCG, underscoring the specificity of our conversion process. By contrast, prior reprogramming efforts—like those described in earlier several studies—often produced mixed endocrine populations, including both β- and α-like cells, due to the shared developmental origins and plasticity among islet cell types. Ensuring such specificity is critical in therapeutic contexts, as inadvertent generation of glucagon-secreting cells may counteract insulin-producing functions. Furthermore, our approach is cell-type agnostic, functioning effectively in human lung endothelial cells, fibroblasts, and PBMCs—making it adaptable for autologous therapies from accessible tissue sources. Overall, this study sets the groundwork for a next-generation, epigenetics-driven platform for efficient, safe, and scalable generation of functional β-cells.
CONCLUSIONS
While these findings are promising, in vivo validation is essential to assess the durability, safety, and functional integration of MEEV-β-derived cells post-transplantation. Additionally, studies on immune compatibility, off-target epigenetic effects, and long-term expression stability will be critical for translational application. Future optimization of delivery methods—e.g., non-viral nanoparticles or mRNA-based systems—may further enhance the clinical feasibility of this platform. Overall, MEEV-β sets the stage for a new generation of precision cell engineering technologies, bridging the gap between synthetic biology and regenerative medicine.
Declaration of competing interest:
The authors have no conflict of interest.
Funding Statement
The authors are thankful to Watch fund grant to RAN by University of Illinois and NIH/NIDCR R01DE027980 to AN for the funding support of this manuscript.
Acknowledgements
We thank the members of our laboratory for their valuable input and critical reading of the manuscript. Their insights greatly contributed to the refinement of this article.
Authorship contribution statement
Raza Ali Naqvi: Conceptualized, written and corrected the manuscript. Amar Singh: Conceptualized, written, edited, and revised the manuscript. Afsar Raza Naqvi: Conceptualized and corrected the final draft. Medha Priyadarshini: Corrected the final draft. Sujata Prasad: Significantly contributed to revision, edited the final draft.
REFERENCES
- Hering, B. J. et al. Phase 3 Trial of Transplantation of Human Islets in Type 1 Diabetes Complicated by Severe Hypoglycemia. Diabetes Care 39, 1230-1240, doi:10.2337/dc15-1988 (2016).
- Shapiro, A. M. et al. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N Engl J Med 343, 230-238, doi:10.1056/NEJM200007273430401 (2000).
- Markmann, J. F. et al. Phase 3 trial of human islet-after-kidney transplantation in type 1 diabetes. Am J Transplant 21, 1477-1492, doi:10.1111/ajt.16174 (2021).
- Hering, B. J. et al. Prolonged diabetes reversal after intraportal xenotransplantation of wild-type porcine islets in immunosuppressed nonhuman primates. Nat Med 12, 301-303, doi:10.1038/nm1369 (2006).
- Graham, M. L. et al. Clinically available immunosuppression averts rejection but not systemic inflammation after porcine islet xenotransplant in cynomolgus macaques. Am J Transplant 22, 745-760, doi:10.1111/ajt.16876 (2022).
- Singh, A. et al. Long-term tolerance of islet allografts in nonhuman primates induced by apoptotic donor leukocytes. Nat Commun 10, 3495, doi:10.1038/s41467-019-11338-y (2019).
- Cardona, K. et al. Long-term survival of neonatal porcine islets in nonhuman primates by targeting costimulation pathways. Nat Med 12, 304-306, doi:10.1038/nm1375 (2006).
- van der Windt, D. J. et al. Long-term controlled normoglycemia in diabetic non-human primates after transplantation with hCD46 transgenic porcine islets. Am J Transplant 9, 2716-2726, doi:10.1111/j.1600-6143.2009.02850.x (2009).
- Naqvi, R. A. et al. The future treatment for type 1 diabetes: Pig islet- or stem cell-derived beta cells? Front Endocrinol (Lausanne) 13, 1001041, doi:10.3389/fendo.2022.1001041 (2022).
- Denner, J. Porcine Endogenous Retroviruses and Xenotransplantation, 2021. Viruses 13, doi:10.3390/v13112156 (2021).
- Millman, J. R. et al. Generation of stem cell-derived beta-cells from patients with type 1 diabetes. Nat Commun 7, 11463, doi:10.1038/ncomms11463 (2016).
- Pagliuca, F. W. et al. Generation of functional human pancreatic beta cells in vitro. Cell 159, 428-439, doi:10.1016/j.cell.2014.09.040 (2014).
- Rezania, A. et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol 32, 1121-1133, doi:10.1038/nbt.3033 (2014).
- Watanabe, T. et al. Evaluating teratoma formation risk of pluripotent stem cell-derived cell therapy products: a consensus recommendation from the Health and Environmental Sciences Institute’s International Cell Therapy Committee. Cytotherapy 27, 1072-1084, doi:10.1016/j.jcyt.2025.04.062 (2025).
- Maxwell, K. G. & Millman, J. R. Applications of iPSC-derived beta cells from patients with diabetes. Cell Rep Med 2, 100238, doi:10.1016/j.xcrm.2021.100238 (2021).
- Pellegrini, S., Zamarian, V. & Sordi, V. Strategies to Improve the Safety of iPSC-Derived beta Cells for beta Cell Replacement in Diabetes. Transpl Int 35, 10575, doi:10.3389/ti.2022.10575 (2022).
- Tyumentseva, M., Tyumentsev, A. & Akimkin, V. CRISPR/Cas9 Landscape: Current State and Future Perspectives. Int J Mol Sci 24, doi:10.3390/ijms242216077 (2023).
- Laurent, M., Geoffroy, M., Pavani, G. & Guiraud, S. CRISPR-Based Gene Therapies: From Preclinical to Clinical Treatments. Cells 13, doi:10.3390/cells13100800 (2024).
- Qi, L. S. et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173-1183, doi:10.1016/j.cell.2013.02.022 (2013).
- Hilton, I. B. et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol 33, 510-517, doi:10.1038/nbt.3199 (2015).
- Riedmayr, L. M. et al. dCas9-VPR-mediated transcriptional activation of functionally equivalent genes for gene therapy. Nat Protoc 17, 781-818, doi:10.1038/s41596-021-00666-3 (2022).
- Yeo, N. C. et al. An enhanced CRISPR repressor for targeted mammalian gene regulation. Nat Methods 15, 611-616, doi:10.1038/s41592-018-0048-5 (2018).
- Liu, G., Lin, Q., Jin, S. & Gao, C. The CRISPR-Cas toolbox and gene editing technologies. Mol Cell 82, 333-347, doi:10.1016/j.molcel.2021.12.002 (2022).
- Nasteska, D. et al. PDX1(LOW) MAFA(LOW) beta-cells contribute to islet function and insulin release. Nat Commun 12, 674, doi:10.1038/s41467-020-20632-z (2021).
- Taylor, B. L., Liu, F. F. & Sander, M. Nkx6.1 is essential for maintaining the functional state of pancreatic beta cells. Cell Rep 4, 1262-1275, doi:10.1016/j.celrep.2013.08.010 (2013).
- Ackermann, A. M., Wang, Z., Schug, J., Naji, A. & Kaestner, K. H. Integration of ATAC-seq and RNA-seq identifies human alpha cell and beta cell signature genes. Mol Metab 5, 233-244, doi:10.1016/j.molmet.2016.01.002 (2016).
- Kang, Y., Kang, J. & Kim, A. Histone H3K4me1 strongly activates the DNase I hypersensitive sites in super-enhancers than those in typical enhancers. Biosci Rep 41, doi:10.1042/BSR20210691 (2021).
- Bell, O., Tiwari, V. K., Thoma, N. H. & Schubeler, D. Determinants and dynamics of genome accessibility. Nat Rev Genet 12, 554-564, doi:10.1038/nrg3017 (2011).
- Dorrell, C. et al. Human islets contain four distinct subtypes of beta cells. Nat Commun 7, 11756, doi:10.1038/ncomms11756 (2016).
- Baumann, K. Stem cells: Insulin-producing beta cells in a dish. Nat Rev Mol Cell Biol 15, 768, doi:10.1038/nrm3907 (2014).
- Kao, C. Y., Mills, J. A., Burke, C. J., Morse, B. & Marques, B. F. Role of Cytokines and Growth Factors in the Manufacturing of iPSC-Derived Allogeneic Cell Therapy Products. Biology (Basel) 12, doi:10.3390/biology12050677 (2023).
- Zhou, Q., Brown, J., Kanarek, A., Rajagopal, J. & Melton, D. A. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature 455, 627-632, doi:10.1038/nature07314 (2008).
- Kalo, E., Read, S. & Ahlenstiel, G. Reprogramming-Evolving Path to Functional Surrogate beta-Cells. Cells 11, doi:10.3390/cells11182813 (2022).
- Niu, F. et al. beta-cell neogenesis: A rising star to rescue diabetes mellitus. J Adv Res 62, 71-89, doi:10.1016/j.jare.2023.10.008 (2024).
- Orive, G. et al. Engineering a Clinically Translatable Bioartificial Pancreas to Treat Type I Diabetes. Trends Biotechnol 36, 445-456, doi:10.1016/j.tibtech.2018.01.007 (2018).
SUPPLEMENTARY MATERIAL
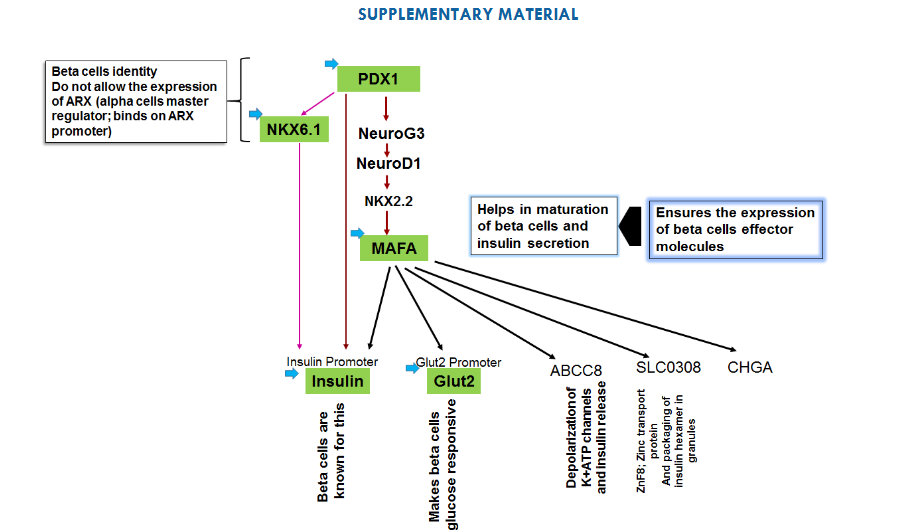
Supplementary Fig. 1: Rationale of choosing PDX1, NKX6.1, insulin, Glut2 and MAFA to induce beta cells. PDX1 expresses in all endocrine cells initially and later on it’s only restricted in beta cells of endocrine pancreas. PDX1 importantly induces the expression of NKX6.1- a transcription factor that restricts the development of beta cells. NKX6.1 reported to bind at the promoter of ARX- a master regulator of alpha cells and also suppresses the glucagon expression. On other hand NKX6.1 also binds at insulin promoter and facilitates the expression of insulin. Thus, NKX6.1 can function as both transcriptional activator as well as transcriptional repressor. Initially, we have thought of taking NKX6.1 to induce the beta cells genetic program, but insulin promoter- the major effector molecule of beta cells not only has NKX6.1 binding site but also PDX1 binding site. Therefore, even though NKX6.1 can be expressed by dCas9.P300core even in the absence of its transcription factor PDX1 but to get the adequate and sustained expression of insulin gene we need to have PDX1 along with NKX6.1. Also, PDX1, induces the genetic program leading to another transcription factor of insulin gene-MAFA, which in turn leads Glut2 gene expression- a glucose transporter. Learning the lessons from stem cells derived beta cells, we came to know the expression of MAFA only possible when PDX1 would induce NKX2.2 via NEUROG3 and NEUROD. Therefore, to reduce the time from PDX1 to MAFA, we have already targeted MAFA, so that it could provide positive feedback for its own expression. Targeting of insulin and Glut2 simultaneously can only enforce the somatic cells to move towards the conversion of beta cells with no further decision-making process related to the beta cells specific genetic network.



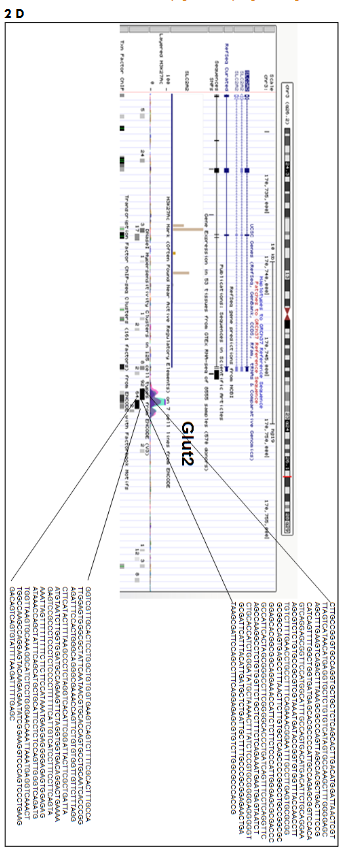

Supplementary Fig. 2: Mapping of DNase I hypersensitive region 5′ to transcription start site (TSS) of β-cells genes using UCSC genome browser. DNA sequence extraction at (A) PDX1, (B) NKX6.1, (C) Insulin, (D) Glut2, (E) MAFA genes for designing of gRNAs.
Source: https://genome.ucsc.edu/cgi-bin/hgGateway?hgsid=782633553_HLl5FvWgiA1juVNq9lmKBHMn9gx3.
Number given along with DNase I hypersensitive site indicate the number of cell line showing this DNase I hypersensitive site.

Supplementary Fig. 3: Snap gene generated maps of (A) pSPgRNA vector (Addgene # 47108) with restriction sites (B) pcDNA-dCas9.P300core (Addgene # 61357). Individual annealed oligonucleotides of each gene were cloned separately in Bbs I site of pSPgRNA vector. All the oligonucleotides (obtained from IDT) were dissolved in nuclease free water to get 100 uM concentration. ~1ul of both 100 uM of forward and reverse oligonucleotides were mixed in 1ul of 10x polynucleotide kinase (PNK) buffer (NEB), 1 ul PNK (NEB) and 4ul nuclease free water. This mixture was kept in thermal cycler with following temperature cycle: 95o C for 5 min, slow cooling till 25o C by reducing 1 C per cycle. Cloning was done as per instructed in step1; Multiplex CRISPR/Cas9 Assembly System Kit protocol (Addgene # 61357). Cut Smart Buffer and Bsb I was obtained from NEB. Each vector containing oligonucleotide sequence was co-transfected in lung endothelial cells along with pc-gRNA-dCads9.p300 (Addgene # 61357). These cells were in culture over a period of 14 days and then RNA was isolated by Rneasy mini kit and SYBR green based qRT-PCR was done to test the best guide.
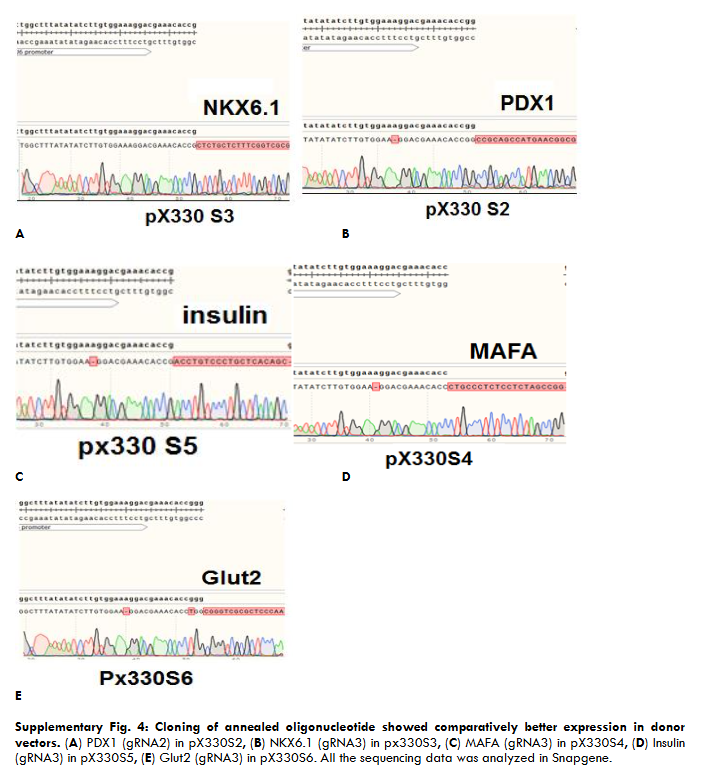
Supplementary Fig. 4: Cloning of annealed oligonucleotide showed comparatively better expression in donor vectors.

Supplementary Fig. 5: Sorting of various Glut2+ cells upon transfection with multiplex epigenetic vector in 14 days. Three experiments were performed per cells, Exp#1, Exp#2, and Exp #3. ~1x 106 cells per transfection were taken into consideration. Gate P3 in (a) and Gate P4 in (B) and (C) depicts the Glut2+ cells.
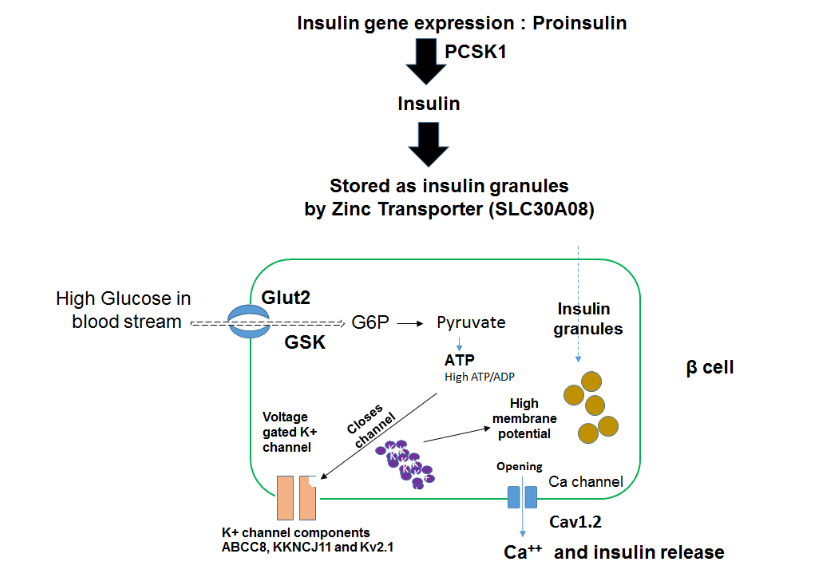
Supplementary Fig. 6: Schematics of genes involved in insulin secretion. The concerted actions of PDX1, NKX6.1, MAFA, NKX2-2, NEUROG1 orchestrates the glucose responsive (Glut2+) β cells in the pancreas to produce insulin. Under the effect of these transcriptional factors, insulin gene transcribes and translates into pro-insulin. PCSK1 converts pro-insulin into mature insulin molecules. SLC30A8 – is Zinc transporter 8 protein, mainly expressed in pancreatic islet cells and mediates zinc (Zn2+) uptake into secretory granules for the storage of insulin molecules. The glucose independent transporter molecule SLC2A2 (also known as GLUT2) transports glucose inside beta cells, which is immediately phosphorylated in glucose 6 phosphate (G6P) by glucokinase (GCK). This reaction is called glycolysis- results in the synthesis of 2 ATP molecules, and results in increased ATP/ADP ratio inside the beta cells. ATP molecule binding with ATP-sensitive K+ (KATP) channels in the beta cell membrane triggers the K+ channel closure. Increase in intracellular K+ ions leads to generation of bursts of action potentials and exocytosis of insulin granules via opening of voltage gated [Ca2+] channels. ABCC8, KKNCJ11 and Kv2.1 are the key components of KATP channels.
SUPPLEMENTARY TABLES
Supplementary Table 1:
List of oligonucleotides DNase I hypersensitive region targeting 5’ to exon 1
| Location | +/- strand | DNase I hypersensitive site score | Chromosomal location of DNase hypersensitive site |
|---|---|---|---|
| PDX1 gRNA 1 -276 to – 298 bp | + | 62 | chr13:28493746-28494295 |
| gRNA2 -521 to – 543 bp | – | 62 | |
| NKX6.1 gRNA1 -107 to -129 bp | + | 77 | chr4:85419146-85420215 |
| gRNA2 -492 to -514 bp | – | 77 | |
| gRNA3 -755 to -777 bp | + | 77 | |
| gRNA4 -349 to -371 bp | – | -95 | chr4:85420226-85420775 |
| Glut2 gRNA1 -127 to -149 bp | – | -92 | chr3:170745821-170746515 |
| gRNA2 -433 to -455 bp | – | -92 | |
| gRNA3 -432 to -454 bp | – | -92 | |
| gRNA4 -1 to -23 bp | + | 64 | chr3:170746526-170747055 |
| Insulin gRNA1 -9 to -31bp | + | 1 | chr11:2184006-2184155 |
| gRNA2 -98 to -120 bp | – | 1 | |
| gRNA3 -96 to -118 bp | + | 1 | |
| gRNA4 -93 to – 115 bp | – | 1 | |
| MAFA gRNA1 -4 to -26 bp | – | -125 | chr8:144512921-144513215 |
| gRNA2 -170 to -192 bp | + | 125 | |
| gRNA3 -90 to -112 bp | + | 57 | chr8:144513626-144514255 |
| gRNA4 -204 to -226 | + | 57 |
Supplementary Table 2:
List of primers for qRT-PCR primers
| Gene | Forward Primer (5’ to 3’) | Reverse Primer (5’ to 3’) | Accession Number | Specificity |
|---|---|---|---|---|
| PDX1 | CCCATGGATGAAGTCTACCAAAGC | AAGTTCAACATGACAGCCAGCT | NM_000209.4 | β-cells |
| NKX6.1 | ACACGAGACCCACTTTTTCCGGA | CTTCTTCCTCCACTTGGTCCGG | NM_006168.2 | β-cells |
| MAFA | CTTCAGCAAGGAGGAGGTCATCC | TCTCCTTGTACAGGTCCCGCT | NM_201589.4 | β-cells |
| Insulin | AGCCTTTGTGAACCAACACCTG | CACGCTTCTGCAGGGACC | NM_001185098.1 | β-cells |
| Glut2 | GCTACCGACAGCCTATTCTAGTGG | TCGCCCTGCCTTCTCCACA | AH002747.2 | β-cells |
| CHGA | CTCTGAACACAGGCAGCTTTCT | ATGTTACAGTCAGGAGTTCTCAGCTTTC | NM_001275.4 | β-cells |
| NeuroD1 | TTGCACCAGCCCTTCCTTTGATG | TCGCTGCAGGATAGTGCATGGTAA | AK313799.1 | β-cells |
| NKX2-2 | TGAACTCTACGCCGTGTTTACAGAATG | GACATTAACGCTGGGACGGTTT | NM_002509.4 | β-cells |
| ABCC8 | ACCACAGCACATGGCTTCATTTC | TGTACAGGTGCAGATGGTGGGATT | NM_001287174.2 | β-cells |
| NEUROG3 | TAAGAGCGAGTTGGCACTGAGCAA | TTTGAGTCAGCGCCCAGATGTAGT | NM_020999.4 | β-cells |
| SLC30A8 | ACAGCCAAGTGGTTCGGAGAGAAA | TTGGGAAACTGACGGTGTGACTGA | NM_173851.3 | β-cells |
| KCNJ11 | GCGCTTTGTGCCCATTGTA | TTGATGGTGTTGCCAAACTTG | NM_000525.3 | β-cells |
| PCSK1 | AGCTGGACCTTCATGTGATACC | GCTAGCCTCTGGATCATAGTTGG | NM_000439.5 | β-cells |
| GAD65 | TTCCTCCAAGCTTGCGTACT | ACCATGCGGAAGAAATTGAC | NM_000818.2 | α-cells |
| CACNA1D | GGTGATCCCCTTCCCCATTC | ATAGTTTGCCTCGTTCGCGT | NM_001128840.3 | β-cells |
| Kv2.2 | AGGGCAGTGTGGGCTCTTC | ATGGTAAATGTCTTGCCTACAGTTGT | NM_004770.2 | β-cells |
| GCK | CTTCCCTCAGTTTTTCGGTGG | TTGATTCCAGCGAGAAAGGTG | NM_001354800.1 | β-cells |
| GCG | AGGCAGACCCACTCAGTGAT | TCGCCCTGCCTTCTCCACA | NM_002054.5 | α-cells |
| GAPDH | CACCAGGGCTGCTTTTAACTCT | GAGGGATCTCGCTCCTGGAAGA | NM_002046.5 | q-RT-PCR control |
Supplementary Table 3:
List of primers for PCR
| Name of gene | Forward primer (5’ to 3’) | Reverse Primer (5’ to 3’) | Purpose |
|---|---|---|---|
| P300 with 50p overlap | TCACCGGCCTGTACGAGACACGG | ATCGACCTGTCTCAGCTGGGAGG | For amplification of P300 |
| Left junction primers | ATCTGGGAGCCCCTGCCG | AGCTGAGGGTCCACAGGTTGACG | For orientation of P300 at left side |
| Right junction primer | GTCGGGATGCGTTTCTCACGCTGG | ATTCTCTTCCCAATCCTCCCCCTTGCTG | For orientation of P300 at right side |
| U6 F | CTAGAGCCATTTGTCTGCAGAATT | To confirm oligonucleotide insertion | |
| CRISPR-step2 | GCCTTTTGCTGGCCTTTTGCTC | CGGGCCATTTACCGTAAGTTATGTAACG | Multiplex assembly |
Supplementary References
1. Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL, Habener JF. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat Genet. 1997 Jan; 15(1):106-10.
2. Sachdeva MM, Claiborn KC, Khoo C, Yang J, Groff DN, Mirmira RG, Stoffers DA. Pdx1 (MODY4) regulates pancreatic beta cell susceptibility to ER stress. Proc Natl Acad Sci U S A. 2009; 106:19090–19095.
3. Daniella A. Babu, Tye G. Deering, Raghavendra G. Mirmira, A feat of metabolic proportions: Pdx1 orchestrates islet development and function in the maintenance of glucose homeostasis, Molecular Genetics and Metabolism,
4. beta-cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the beta-cell phenotype and maturity onset diabetes. Volume 92, Issues 1–2, 2007, Pages 43-55,
5. Ahlgren, U. et al. Genes Dev. 1998 Jun 15;12(12):1763-8.
6. Ashleigh E. Schaffer, Brandon L. Taylor, Jacqueline R. Benthuysen, Jingxuan Liu, Fabrizio Thorel, Weiping Yuan, Yang Jiao, Klaus H. Kaestner, Pedro L. Herrera, Mark A. Magnuson, Catherine Lee May, Maike Sander. Nkx6.1 Controls a Gene Regulatory Network Required for Establishing and Maintaining Pancreatic Beta Cell Identity. PLoS Genet. 2013 Jan; 9(1): e1003274.
7. Schisler J. C. et al. The Nkx6.1 homeodomain transcription factor suppresses glucagon expression and regulates glucose-stimulated insulin secretion in islet beta cells. Proc Natl Acad Sci U S A. 2005;102(20):7297–7302.
8. Brandon L. Taylor, Fen-Fen Liu, Maike Sander. Nkx6.1 is essential for maintaining the functional state of pancreatic beta cells. Cell Rep. Author manuscript; available in PMC 2014 Jun 15. Published in final edited form as: Cell Rep. 2013 Sep 26; 4(6): 1262–1275.
9. Fu Z, Gilbert ER, Liu D. Regulation of insulin synthesis and secretion and pancreatic Beta-cell dysfunction in diabetes. Curr Diabetes Rev. 2013;9(1):25–53.
10. Waeber G., Thompson N., Haefliger J.A., Nicod P. Characterization of the murine high Km glucose transporter GLUT2 gene and its transcriptional regulation by glucose in a differentiated insulin-secreting cell line. J. Biol. Chem. 1994; 269:26912–26919.
11. Leturque A., Brot-Laroche E., Le Gall M. GLUT2 mutations, translocation, and receptor function in diet sugar managing. Am. J. Physiol. Endocrinol. Metab. 2009;296: E985–E992.
12. Benzinou M, Creemers JW, Choquet H, et al. Common nonsynonymous variants in PCSK1 confer risk of obesity. Nat Genet. 2008;40(8):943–945.
13. Nicolson TJ et al. Diabetes. 2009 Sep; 58(9):2070-83.
14. Frans C. et al. Diabetes Jan 2001, 50 (1) 1-11.
15. Matschinsky, F.M. et al., 1968. Metabolism of glucose in the islets of Langerhans. J. Biol. Chem. 243:2730-2736
16. Yasuo T, et al., J Clin Invest. 2007;117(1):246-257
17. Li XN et al., J Pharmacol Exp Ther. 2013 Feb;344(2):407-16.
18. Frances M. et al., Nat Rev Endocrinol. 2013 Nov; 9(11): 660–669.