





Growth Factors and Pancreatic Hormones in NAFLD
Possible roles of growth factors and pancreatic hormones in the physiopathology of the non-alcoholic fatty liver disease
Martha Lucinda Contreras-Zentella1, Lorena Carmina Hernández-Espinosa1, and Rolando Hernández-Muñoz1*
- Department of Cell Biology and Development, Institute de Cellular Physiology, Universidad Nacional Autónoma de México (UNAM). México City 04510, Mexico.
OPEN ACCESS
PUBLISHED 31 October 2025
CITATION Contreras-Zentella, ML., Hernández-Espinosa, LC., and Hernández-Muñoz, R., 2025. Possible roles of growth factors and pancreatic hormones in the physiopathology of the non-alcoholic fatty liver disease. Medical Research Archives, [online] 13(10). https://doi.org/10.18103/mra.v13i10.7013
COPYRIGHT: © 2025 European Society of Medicine. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
DOI https://doi.org/10.18103/mra.v13i10.7013
ISSN 2375-1924
ABSTRACT
The prevalence of nonalcoholic fatty liver disease (NAFLD) has increased paralleling with diabetes and metabolic syndrome. This trend has contributed to a surge in metabolic dysfunction–associated steatotic liver disease (MASLD), the most common chronic liver disease worldwide. NAFLD is defined by excessive triglycerides (TG) accumulation in hepatocytes, independent of alcohol consumption. Fatty liver arises from either increased TG delivery to the liver or carbohydrate conversion into TG. NAFLD shares key pathogenic mechanisms with metabolic syndrome, including obesity, hyperlipidemia, insulin resistance, mitochondrial dysfunction, oxidative stress, and inflammation. Two growth factors—Hepatocyte growth factor (HGF) and Epidermal growth factor (EGF)—may play significant roles in NAFLD pathophysiology. HGF interacts with insulin to regulate glucose metabolism in hepatocytes contributing to glucose homeostasis. EGF may also influence glucagon secretion by counteracting its suppression by plasma glucose, besides to be involved in liver cell proliferation. The known effects of these growth factors and hormones are largely loss at the onset of NAFLD. Therefore, this review explores the potential roles of HGF, EGF, and the pancreatic hormones insulin and glucagon in the NAFLD and evaluates their possible utility as biomarkers for diagnosis and therapeutic monitoring, since NAFLD, as an emerging disease, requires early detection and accurate prediction of disease progression for effective management.
Keywords: Insulin; Glucagon; Triacylglycerols; Oxidant stress; Inflammation; FFA.
Introduction
Nonalcoholic fatty liver disease (NAFLD) is a chronic liver disorder that occurs in individuals with minimal or no alcohol consumption and has an estimated global adult prevalence of approximately 25%1. Over recent decades, NAFLD has emerged as one of the most common chronic liver diseases worldwide, with the potential to progress to cirrhosis and hepatocellular carcinoma2. More recently, the terminology has shifted to metabolic dysfunction–associated steatotic liver disease (MASLD) and metabolic dysfunction–associated steatohepatitis, designations that encompass a spectrum of conditions characterized by excessive lipid accumulation in hepatocytes, leading to inflammation, fibrosis, and cirrhosis, in the absence of significant alcohol intake3,4.
The prevalence of MASLD continues to rise, particularly among individuals with obesity, where intrahepatic triglyceride (TG) accumulation can drive disease progression from simple steatosis to MASH and ultimately to advanced fibrosis and cirrhosis5,6. Patients with MASLD frequently present with metabolic comorbidities, including hypertension, cardiovascular disease, insulin resistance, and type 2 diabetes mellitus (T2DM)7. Lipid accumulation is closely associated with insulin resistance in MASLD pathogenesis. Pro-inflammatory M1 macrophages in hypertrophic adipose tissue secrete a variety of chemokines and cytokines8, including monocyte chemoattractant protein-1, tumor necrosis factor–α, and interleukins IL-6, IL-2, and IL-8.
Disease progression is driven by both well-established mechanisms—chronic overnutrition, adipose tissue dysfunction, insulin resistance, and substrate overload–induced hepatic lipotoxicity—and less fully elucidated processes such as hepatocellular stress pathways, immune cell recruitment, cytokine and interleukin signaling, gut dysbiosis, and fibrogenic activation9. The rising prevalence of metabolic syndrome, fueled by increasing rates of obesity and T2DM, is a major contributor to the burden of chronic hepatic steatosis, with significant implications for healthcare systems and socio-economic resources.
Early and accurate diagnosis is essential for effective management. Although liver biopsy remains the diagnostic gold standard, it is invasive, carries procedural risks, and is limited by sampling error and interobserver variability. Advances in medical technology, alongside growing emphasis on patient-centered care, have accelerated the development and application of non-invasive tests. In early-stage or asymptomatic diseases, non-invasive tests improve diagnostic accuracy while reducing reliance on biopsy, thereby minimizing patient discomfort and lowering healthcare costs. Despite the availability of numerous non-invasive tests for NAFLD, their widespread clinical adoption remains restricted due to technical, logistical, and standardization challenges10–12. On the other hand, two important growth factors, hepatocyte growth factor (HGF) and epidermal growth factor (EGF), may play a significant role in the pathophysiology of NAFLD.
HGF acts as a ligand for the MET tyrosine kinase receptor (c-MET), activating the MAPK and PI3K/AKT signaling pathways13. Disruption of hepatocyte c-MET (cellular mesenchymal–epithelial transition) signaling triggers chemotactic and inflammatory responses, underscoring the anti-inflammatory properties of HGF14. In T2DM, the HGF–MET system improves insulin sensitivity in murine models of insulin resistance, likely through direct interactions between the MET receptor and the insulin receptor, thereby influencing hepatic glucose metabolism; moreover, c-MET expression in β-cells enhances HGF sensitivity, impacting cell growth, survival, and insulin secretion15. The EGF/EGF receptor (EGF/EGFR) signaling pathway plays a pivotal role in regulating cell growth, proliferation, migration, and differentiation, both in physiological conditions and in pathological contexts such as cancer and T2DM-related cardiovascular dysfunction16. Administration of EGF and gastrin to alloxan-induced diabetic mice rapidly normalized blood glucose levels within a few days, supporting a role for EGF in modulating insulin sensitivity17.
Recently, we reported that insulin and glucagon regulate serum glucose and lipid levels, with HGF and EGF concentrations varying according to sex. This association was particularly evident in T2DM, where impaired cell proliferation or repair mechanisms contribute to metabolic disturbances18.
Based on these observations, the present study aimed to investigate the potential correlations between serum HGF, EGF, insulin, and glucagon with glucose, lipid profile, and key liver function parameters in patients with NAFLD/MASLD, and to assess the potential diagnostic utility of these proteins.
Hepatocyte growth factor and the liver
Hepatocyte growth factor is a secreted protein originally identified as a growth factor that is ubiquitously present in the extracellular matrix of most tissues19. It has a heterodimeric structure composed of a heavy chain of approximately 60 kDa and a light chain of about 3.5 kDa. HGF is a cytokine expressed and produced by various mesenchymal cell types, such as hepatic stellate cells, vascular endothelial cells, and Kupffer cells. Its receptor, c-MET, dimerizes and activates receptor tyrosine kinase (RTK) activity in the intracellular domain, mediating HGF action20,21.
The α-chain (heavy) contains an N-terminal hairpin domain and four kringle domains (K1–K4). The first two kringle domains and the hairpin domain are essential for HGF to perform its biological functions. The β-chain contains a serine protease homology domain, which is the binding site for c-MET. Serine proteases such as matriptase and HGF activator (HGFA) are involved in the proteolytic processing of HGF22,23.
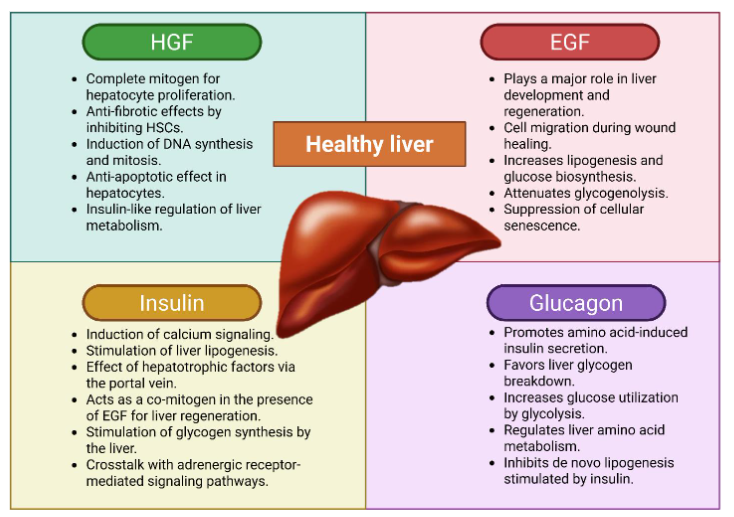
Upon binding to c-MET, HGF triggers the receptor’s intrinsic kinase activity, activating downstream cascades such as JAK/STAT3, PI3K/NF-κB, Akt, and Ras/Raf pathways. These pathways regulate cell proliferation, growth, and survival24. c-MET is expressed on the surface of epithelial cells in multiple organs, including the liver, pancreas, prostate, kidney, lung, and bronchus25. Although initially identified and cloned as a potent mitogen for hepatocytes, HGF is now recognized as a protective and trophic factor for many tissues and organs. For example, it directly promotes proliferation and differentiation of erythroid progenitors26 and plays an important role in wound healing and tissue repair, acting as an antifibrotic agent in the kidney, heart, and lung27,28.
Conversely, overactivation of the HGF/c-MET pathway has been implicated in the pathogenesis and prognosis of multiple neoplastic conditions. c-MET is a proto-oncogene, and its constitutively active mutant form was originally cloned as a transforming factor from a chemically induced human osteosarcoma cell line29.
Regarding the liver, this organ has the remarkable ability to regenerate to its optimal volume after resection30. HGF is one of only two complete mitogens—alongside epidermal growth factor receptor (EGFR)—that induce hepatocyte DNA synthesis and mitosis. Activation of the HGF/c-MET axis initiates intracellular signaling cascades that drive G1–S progression in hepatocytes. Evidence shows that following various liver injuries, such as partial hepatectomy, ischemia, or hepatitis, there is intense extracellular matrix remodeling, increased protease activity, and robust intracellular signaling31. Active HGF levels rise rapidly in the liver due to increased production by Kupffer cells, stellate cells, and sinusoidal endothelial cells, followed by activation via urokinases32. Since plasma HGF levels also increase, an endocrine mechanism has been proposed. Moreover, HGF transcript levels and activity are markedly elevated in intact organs such as the lung, kidney, and spleen after liver injury33. These changes collectively support balanced liver growth and regeneration.
During the proliferation phase, HGF induces hepatocyte DNA synthesis and mitosis as a complete mitogen. In PH models in rats, c-MET receptors are highly activated within 0.5 h after PH, while HGF levels initially decrease in the first 0–3 h, then rise significantly between 3-48 h before being inactivated in the termination phase through interaction with TGF-β34,35. The c-MET-deficient mice show increased mortality after 70% partial hepatectomy36, highlighting the essential role of the HGF/c-MET axis in liver regeneration.
Preclinical data indicate that HGF is a promising therapeutic tool for liver diseases. Strategies including gene therapy, mesenchymal cell transfection to overexpress HGF, and recombinant HGF administration have shown beneficial effects. In experimental rat models, HGF suppressed the development of liver cirrhosis after toxic injury37,38 and cholestatic damage, prevented liver failure, and—while exerting anti-apoptotic effects on hepatocytes39—induced pro-apoptotic and inhibitory effects on hepatocellular carcinoma cells40.
Epidermal growth factor and the liver
The EGF is a member of the growth factor family, and the activation of the EGF/EGFR axis has been extensively studied in various biological processes. In the liver, EGF is involved in multiple pathological conditions, including inflammation, fibrosis, and tumor metastasis41,42. Through its receptor, EGF acts as a potent mitogen, promoting cellular proliferation and differentiation in hepatocytes43.
EGF is a 53–amino acid protein, approximately 6 kDa in molecular weight, containing three disulfide bonds. It binds to EGFR, activating intracellular signaling pathways via phosphorylation of the receptor tyrosine kinase (RTK). In vivo, EGFR phosphorylation occurs 30–60 minutes after PH in rodents32. EGFR, a membrane protein found in epithelial and mesenchymal cells, belongs to the ErbB family, which also includes HER2/ErbB2/c-neu, HER3/ErbB3, and HER4/ErbB4. EGFR can bind seven known ligands, including EGF and transforming growth factor-α. Ligand binding triggers receptor dimerization, autophosphorylation, and activation of multiple intracellular signaling pathways44,45.
The EGFR structure consists of three regions: an extracellular domain with four subdomains responsible for ligand binding and dimerization, a single hydrophobic transmembrane domain, and an intracellular kinase domain46. Upon ligand binding, EGFR undergoes dimerization and autophosphorylation of tyrosine residues in the C-terminal cytoplasmic region, enabling the recruitment of adaptor proteins containing phosphotyrosine-binding Src homology 2 domains32.
EGFR signaling activation depends on the ligand type and binding affinity, leading to either stable or transient homodimer formation or heterodimerization with other ErbB receptors. High-affinity ligands include EGF, transforming growth factor-α, HB-EGF, and BTC, while AREG, EREG, and EPGN are low-affinity ligands. Recent bioinformatic, molecular dynamics, and crystallographic analyses have revealed that certain ligands induce distinct conformations of the receptor. For instance, EREG and EPGN promote fewer stable dimers yet sustain EGFR and ERK activation over time32,47.
EGFR can also be activated through G protein-coupled receptor-mediated transactivation, involving two main mechanisms. The first relies on metalloproteases, particularly the ADAM family, which release membrane-bound EGFR ligands into the extracellular space. The second involves the P2Y2 receptor, a purinergic G protein-coupled receptor, which upon ATP binding, enhances ERK phosphorylation and stimulates cell proliferation32,48.
EGF is crucial for liver development and regeneration, with EGFR activation being essential for physiological liver growth. Ligand binding initiates signaling cascades primarily driving hepatocyte proliferation rather than survival after liver injury. This has been confirmed in EGFR knockout models, where no difference in apoptosis was observed compared with wild-type mice49.
EGF activates multiple intracellular signaling networks, including the RAS–RAF–MEK–MAPK pathway, PI3K–Akt pathway, and JAK–STAT cascade50. These pathways mediate diverse biological effects, such as suppressing cellular senescence to promote proliferation, stimulating epithelial and fibroblast migration to injury sites, modifying the extracellular matrix, inducing myofibroblast proliferation, enhancing re-epithelialization, promoting keratinocyte motility, triggering angiogenesis, facilitating adipocyte maturation, regulating thymocyte differentiation, supporting mammary gland lactogenesis, and modulating metabolism, including lipogenesis and biosynthesis of glucose and amino acids51,52.
EGF also stimulates hepatic stellate cell growth, contributing to liver fibrosis53. Clinically, elevated serum EGF levels have been observed in patients with hepatocellular carcinoma and chronic hepatitis C compared to those with hepatitis C alone or health controls, suggesting a role in virus-associated oncogenesis54. Beyond proliferation, EGF influences quiescent cell functions and interacts with hormones to regulate metabolism. Notably, EGF attenuates the glycogenolytic effects of glucagon55 and catecholamines56 and interferes with insulin-stimulated glycogen storage in adult rat hepatocytes57.
Insulin and the liver
Human insulin is a heterodimeric protein composed of an A chain with 21 amino acids and a B chain with 30 amino acids, connected by two disulfide bonds. It is synthesized by pancreatic β-cells and reaches the liver through the portal circulation. The insulin receptor is a built-in tyrosine kinase receptor, composed of two extracellular α-subunits (which bind insulin) and two transmembrane β-subunits (which possess tyrosine kinase activity)58. Upon insulin binding, the receptor autophosphorylates and phosphorylates intracellular substrates such as insulin receptor substrate-1. This initiates downstream signaling cascades involving phosphoinositide 3-kinase (PI3K) and protein kinase B (Akt), ultimately promoting the translocation of glucose transporter-4 to the cell surface to facilitate glucose uptake59.
Insulin’s main physiological role is to lower blood glucose levels by stimulating cellular glucose uptake and promoting the synthesis of glycogen, fat, and protein60. Interestingly, EGF and insulin share many biological activities, including stimulation of cell proliferation61, regulation of ion and glucose fluxes62, enhancement of glycolysis63, and promotion of fatty acid and glycogen synthesis64, largely through activation of receptor-linked tyrosine kinase signaling65. EGF may also influence insulin secretion via receptors on β-cells or reduce plasma glucose levels, indicating its potential as a pharmacological agent for T2DM.
In vitro, insulin and EGF are essential for hepatocyte survival and proliferation in culture systems. Their intracellular signaling often acts indirectly as mitogens by inducing the secretion of autocrine growth factors such as transforming growth factor-alpha and insulin-like growth factor-I59. It has long been established that insulin contributes to liver regeneration and functions as a key metabolic regulator during hepatocyte proliferation66,67. Insulin in the portal vein acts as a hepatotrophic factor68, and increased insulin-binding sites on hepatocyte membranes have been observed 24 to 48 hours after 70% partial hepatectomy in rats, indicating a close association between insulin and liver regeneration69. While insulin alone can promote DNA synthesis and hepatocyte proliferation, it is often described as a co-mitogen that enhances the effects of direct mitogens such as EGF. Signaling pathways involving receptor tyrosine kinase, PI3K, extracellular signal-regulated kinase (ERK), and mammalian target of rapamycin (mTOR) mediate these proliferative effects70,71.
Pharmacological activation of insulin/insulin growth factor-1 signaling and nuclear factor erythroid 2–related factor 2 (Nrf2) pathways represent promising strategies to improve liver regeneration in acute or chronic liver injury59.
Further studies revealed that insulin exposure elevates nuclear and cytosolic levels of inositol 1,4,5-trisphosphate–mediated calcium signals. Experiments buffering nuclear IP3 demonstrated a significant reduction in bromodeoxyuridine uptake, confirming that insulin-induced hepatocyte proliferation depends on nuclear IP3 formation in vivo60. Mechanistically, insulin binding to its receptor triggers receptor internalization and translocation to the nucleus, where it activates phospholipase C. Before insulin stimulation, insulin receptors predominantly localize to the plasma membrane; within 5 minutes of insulin exposure, they are redistributed inside the nucleus and cytoplasm72.
Insulin also regulates lipogenesis in hepatocytes by increasing transcription of lipogenic enzymes via enhanced nuclear translocation of transcription factors such as carbohydrate-responsive element-binding protein and sterol regulatory element-binding protein-1c. These factors synergistically promote expression of lipogenic genes in the presence of glucose and insulin. In contrast, glucagon—via cAMP—and polyunsaturated fatty acids inhibit carbohydrate-responsive element-binding protein and sterol regulatory element-binding protein-1c activities, respectively73.
Glucagon and the liver
Glucagon is a 29–amino acid peptide hormone secreted by pancreatic α-cells. It is derived from the precursor proglucagon, which can be processed into several related peptide hormones expressed in pancreatic islet α-cells, intestinal enteroendocrine L cells, and, to a lesser extent, neurons in the brainstem and hypothalamus75. The processing of proglucagon is mediated by the enzyme’s prohormone convertase and prohormone convertase76.
Glucagon exerts effects on various organs, including the liver, pancreas, kidney, and brain77. Its role in hepatic glucose production via glycogenolysis and gluconeogenesis is well characterized. Glucagon stimulates glycogen breakdown in a dose-dependent manner mediated through cAMP, as well as inducing transcriptional changes in key gluconeogenic enzymes like phosphoenolpyruvate carboxykinase. Notably, glucagon receptors are absent in human muscle cells and adipocytes; thus, glucagon’s effect on gluconeogenesis likely depends on substrate availability mediated by lipolysis and/or proteolysis77,78.
Glucagon is fundamental to cellular homeostasis, and its secretion is intricately regulated in coordination with insulin secretion, which also reciprocally regulates glucagon release. Dysregulation of this balance contributes to hyperglucagonemia, which has been implicated in hyperglycemia associated with T2DM79.
Stimulation of pancreatic β-cells via glucagon binding to glucagon receptors, as well as to glucagon-like peptide 1 receptors, is crucial for amino acid–induced insulin secretion. The pancreas–liver interplay, termed the liver–alpha cell axis, highlights glucagon’s importance in glucose, amino acid, and lipid metabolism. In patients with fatty liver disease, this axis can be disrupted, leading to elevated glucagonotropic amino acids, dyslipidemia, and hyperglucagonemia—a condition recently described as “glucagon resistance”80.
Therapeutic use of glucagon and related agents has spurred interest in understanding its mechanisms and pharmaceutical potential in fatty liver disease. Research into glucagon signaling has led to the development of several pharmacological agents, including glucagon receptor antagonists, agonists, and novel dual or triple receptor agonists that combine glucagon and incretin hormone receptor activities. These advances leverage glucagon’s central role in cellular homeostasis to therapeutically restore glycemic control80. Alpha cell function also depends on the regulatory input from pancreatic β-cells and somatostatin-producing delta cells79,80.
Hyperglucagonemia is frequently observed in individuals with NAFLD, independent of type 2 diabetes status. A key question remains whether obesity, type 2 diabetes, or NAFLD primarily drives hyperglucagonemia85. Preliminary data from our laboratories suggest that obesity and NAFLD may induce hyperglucagonemia via distinct mechanisms, potentially related to elevated amino acid and fatty acid levels that disrupt the liver–alpha cell axis, as seen in NAFLD86. Other studies suggest that hyperglucagonemia correlates more strongly with hepatic steatosis than with diabetes per se85. Individuals with NAFLD appear to exhibit glucagon resistance, impairing the liver–alpha cell axis and resulting in hyperglucagonemia and hyperaminoacidemia87.
Clinical trials have demonstrated that co-agonists targeting glucagon-like peptide 1 and glucagon receptors, as well as triple agonists targeting glucagon-like peptide 1 receptors/GIP/glucagon receptors, effectively regulate energy metabolism and show promise in treating nonalcoholic steatohepatitis88,89. The development of glucagon-based therapeutics, especially in combination with glucagon-like peptide 1 receptor agonists, represents a significant advance for metabolic liver diseases commonly associated with obesity and T2DM77,90.
Non-alcoholic fatty liver disease (NAFLD): underlying mechanisms
NAFLD is primarily characterized by the accumulation of lipid droplets within hepatocytes (steatosis), which is commonly driven by insulin resistance and metabolic syndrome. This lipid buildup induces hepatocyte stress, lipid peroxidation, and inflammation, which may progress to non-alcoholic steatohepatitis91. The spectrum of NAFLD ranges from simple hepatic TG accumulation (simple steatosis) to metabolic dysfunction-associated steatohepatitis, hepatic fibrosis, and cirrhosis. It represents the most prevalent liver disease worldwide and its incidence rises in parallel with obesity and T2DM92. Central to NAFLD/MASLD pathogenesis is the dysregulation of hepatic fatty acid metabolism, disrupting the balance between free fatty acid oxidation and lipid storage. The classic “two-hit hypothesis” describes NAFLD development as: (a) excessive hepatic lipid deposition and (b) subsequent activation of inflammatory cascades, oxidative stress, and fibrogenesis93.
The majority (~60%) of hepatic TGs originate from increased lipolysis of insulin-resistant adipose tissue due to impaired insulin signaling and elevated hormone-sensitive lipase expression, releasing FFAs into circulation94-97. The remaining ~25% derive from de novo lipogenesis, where excess carbohydrates are converted into free fatty acids in the liver94. De novo lipogenesis is regulated by nuclear transcription factors, chiefly sterol regulatory element-binding protein-1c and carbohydrate-responsive element-binding protein, which enhance expression and activity of lipogenic enzymes such as acetyl-CoA carboxylase, fatty acid synthase, and stearoyl-CoA desaturases98.
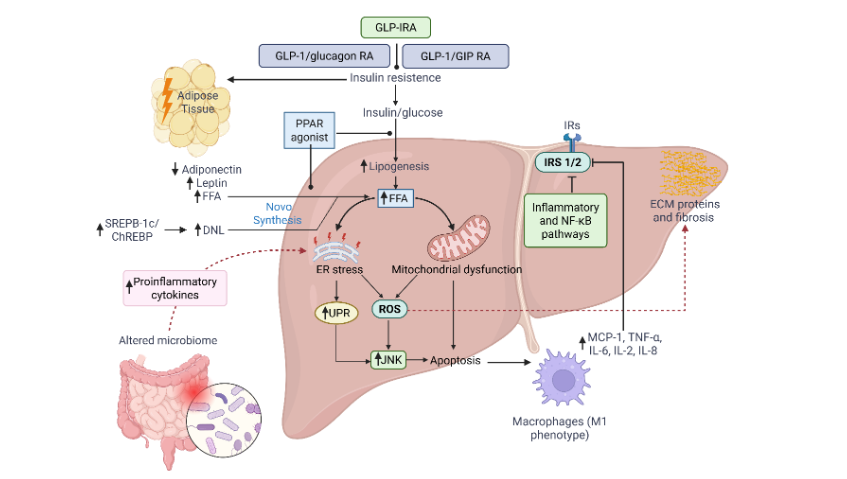
In MASLD progression, M1 macrophages in expanded adipose tissue secrete pro-inflammatory chemokines and cytokines including monocyte chemoattractant protein-1, tumor necrosis factor–alpha, and interleukins IL-6, IL-2, and IL-88. TNF-α induces serine/threonine phosphorylation of insulin receptor substrate-1, impairing its insulin signaling capacity and promoting insulin resistance99.
NASH, an advanced NAFLD stage, is characterized by hepatic inflammation, cellular damage, mitochondrial dysfunction, and oxidative stress. Its progression fits the “two-hit” model where initial steatosis (first hit) is followed by inflammatory and oxidative injury (second hit). Pro-inflammatory cytokines IL-6, IL-1β, and tumor necrosis factor-α are key mediators of inflammation and fibrosis in NASH100. Recent research highlights mitochondrial dysfunction and reactive oxygen species overproduction as central in NASH pathogenesis, along with altered gut microbiota that amplifies hepatic inflammatory signaling101.
Emerging evidence implicates inflammatory pathways in insulin resistance development. The gut microbiome, significantly altered in NAFLD, may be an early contributor to IR and adipose tissue dysfunction96. Hepatic IR involves inflammatory and NF-κB signaling, where the IκB kinase (IKK)–NF-κB axis plays a pivotal role by activating NF-κB through phosphorylation and inactivation of its inhibitor IκB97,102. Insulin receptor substrate-1 and -2 are critical insulin signaling mediators in the liver, with their impairment by inflammatory cytokines, a fundamental mechanism driving insulin resistance103.
Oxidative stress is a hallmark of NAFLD. Nutrient overload stresses the hepatic mitochondrial electron transport chain, generating reactive species such as superoxide, hydroxyl radicals, hydrogen peroxide, and nitric oxide. Reactive nitrogen species also increase due to aberrant protein catabolism, exacerbating cellular oxidative stress104. Obese livers show elevated fatty acid oxidation without proportional increases in fatty acid uptake or esterification compared to lean counterparts105. Hepatic sources of reactive oxygen species include mitochondria, endoplasmic reticulum, xanthine oxidase, peroxisomes, and cytochrome P450 enzymes. Antioxidant defenses—superoxide dismutase, catalase, and glutathione peroxidase—normally neutralize ROS, but in MASH, increased reactive oxygen species production alongside impaired antioxidant capacity drives disease progression106,107. Reactive oxygen species react with polyunsaturated fatty acids, causing lipid peroxidation and generating toxic aldehydes like malondialdehyde and 4-hydroxynonenal108. These lipid peroxidation products activate hepatic stellate cells, promoting extracellular matrix deposition and fibrosis109. In MASH, diminished hepatic glutathione and reduced activities of antioxidant enzymes correlate with severity110.
Substrate overload-induced oxidative stress and mitochondrial dysfunction stimulate hepatic pro-inflammatory cytokine release, advancing MASLD111. Mitochondrial injury triggers mitophagy, a selective autophagy process; impaired mitophagy in MASH leads to inflammasome activation (Figure 2). Elevated expression of methylation-controlled J protein, a mitochondrial inner membrane protein that inhibits respiratory chain complex I, is observed in MASH livers. Reduced methylation-controlled J protein enhances fatty acid oxidation, mitigating lipid accumulation and hepatocyte injury112-114.
Macrophage infiltration in visceral adipose tissue fosters a pro-inflammatory milieu, exacerbating insulin resistance—a key driver in MASLD pathogenesis115,116. Insulin resistance disrupts normal lipolysis and exacerbates de novo lipogenesis, exceeding hepatic metabolic capacity and generating lipotoxic species that induce oxidative and endoplasmic reticulum stress, activate inflammasomes, and cause hepatocyte apoptosis117,118. Autophagy, a critical intracellular recycling process, facilitates lipolysis and free fatty acid release under starvation, mitigating NAFLD progression through lipophagy119,120.
Human microbiome studies reveal specific microbial signatures linked to NAFLD severity, supporting the gut–liver axis as a therapeutic target to reduce liver inflammation and metabolic dysfunction121. Overall, MASLD pathophysiology is complex and multifactorial. Despite advances, many underlying mechanisms remain incompletely understood100,122.
Role of growth factors and pancreatic hormones during NAFLD
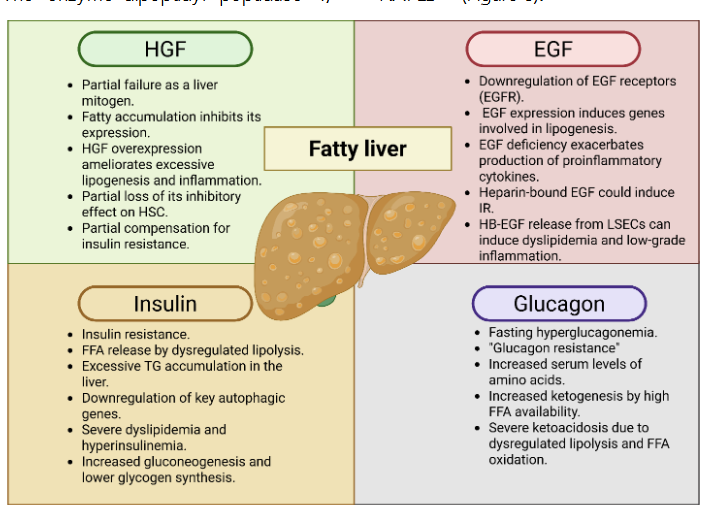
Insulin resistance, oxidative stress, and metabolic disturbances represent key pathological factors in the onset of NAFLD and its progression to fibrosis. Current therapeutic approaches and investigational drugs for NAFLD-related fibrosis primarily aim to reduce IR and correct metabolic abnormalities, thereby decreasing free fatty acid production, lipotoxicity, and excessive TGs accumulation in hepatocytes, mitochondrial dysfunction, and endoplasmic reticulum stress123. The insulin resistance is a hallmark of NAFLD pathogenesis124, characterized by impaired glucose uptake in peripheral tissues such as adipose tissue and muscle125. Insulin resistance in adipose tissue promotes dysregulated lipolysis and inappropriate free fatty acid release, which exacerbates systemic insulin signaling defects and highlights the metabolic crosstalk between adipose tissue and the liver. Adipose-derived factors such as adiponectin, IL-6, and other peptides exert both protective and proinflammatory effects on hepatic tissue126. The enzyme dipeptidyl peptidase 4, secreted by hepatocytes, may further contribute to IR, linking hepatic and adipose tissue dysfunction127.
Lipolysis and autophagy share regulatory mechanisms, being activated under nutrient-deprived conditions and modulated by hormonal signals—stimulated by glucagon and inhibited by insulin128. Mouse models of diabetes and obesity demonstrate that hyperinsulinemia and insulin resistance correlate with autophagy defects, attributed to downregulation of key autophagic genes129. According to Tilg et al86, both hepatic and peripheral insulin resistance worsen with NAFLD progression. IR involves decreased glucose uptake due to impaired insulin signaling, increased gluconeogenesis, and reduced hepatic glycogen synthesis. Obese individuals with T2DM exhibit more pronounced dyslipidemia, hyperinsulinemia, and IR in hepatic and adipose tissues than non-obese individuals without NAFLD130.
Tumor necrosis factor-α promotes serine/threonine phosphorylation of insulin receptor substrate 1, impairing insulin signaling and inducing insulin resistance131. Additionally, hyperinsulinemia and hyperglycemia directly contribute to oxidative stress132. Liver-specific downregulation of JMJD3, a histone H3K27 demethylase, results in mitochondrial β-oxidation defects and autophagy deficiencies, leading to TG accumulation, steatosis, and glucose intolerance—factors critical in MASLD progression133. Metabolic stressors such as high-fat diets and IR reduce sirtuin-1 activity; conversely, endogenous activators like NAD+ and pharmacological agents such as sibutramine can enhance it134. Sirtuin-2 upregulation in obese mice alleviates insulin resistance, steatosis, and inflammation, while sirtuin-2 knockdown exacerbates metabolic dysfunction135. AKT phosphorylation correlates with IR and apoptosis in fatty liver disease, and mitochondrial pyruvate carrier 1 has been linked to inflammation, fibrosis, and insulin sensitivity in murine NAFLD/MASH models134.
The incretin glucose-dependent insulinotropic polypeptide regulates glucose homeostasis by modulating insulin and glucagon secretion based on glycemic status136, positioning it as a promising therapeutic target for NAFLD/NASH given the metabolic overlap with type 2 diabetes. Fasting glucagon levels may contribute to NAFLD development, with hepatic IR proposed as a determinant of fasting hyperglucagonemia independent of diabetes status85,137. Glucagon levels correlate with plasma amino acid concentrations85, and amino acids such as glutamine and alanine exert strong glucagonotropic effects, modulating hepatic amino acid metabolism138. Pancreatic clamp studies show glucagon’s impaired regulation of amino acid metabolism in obese versus lean subjects139.
NAFLD not only drives IR but also hepatic glucagon resistance, resulting in elevated circulating amino acids that stimulate α-cell glucagon secretion140. Patients with MASLD exhibit glucagon resistance, hyperglucagonemia, and hyperaminoacidemia77,85,137,141. Glucagon resistance towards hepatic lipid metabolism is implicated in NAFLD pathophysiology142. Glucagon-mediated free fatty acid elevation enhances ketogenesis, and suppression of glucagon secretion by somatostatin may prevent ketoacidosis development in T1DM143.
NAFLD progression spans from simple steatosis to non-alcoholic steatohepatitis, characterized by steatosis, hepatocyte apoptosis, inflammation, and fibrosis. Assessing the impact of steatosis on liver regeneration is complicated by concurrent fibrosis during disease advancement144. TG accumulation in hepatocytes correlates with impaired liver volumetry and regeneration145, although mechanisms underlying hepatocyte proliferation defects or replicative senescence remain unclear. Metabolic dysfunction-associated steatotic liver disease (MASLD) and liver regeneration share critical pathways—including HGF/c-Met, EGFR, Wnt/β-catenin, and Hippo/YAP-TAZ signaling—all of which are disrupted in MASLD. EGFR expression is downregulated in obese mouse models with MASH, but growth hormone administration rescues hepatocyte proliferation and restores EGFR expression, improving liver regeneration146. Paradoxically, EGFR inhibitors alleviate steatosis, inflammation, and fibrosis in MAFLD mouse models147.
Exacerbated cytokine production148, EGFR pathway deficiencies146, and oxidative stress149 contribute to impaired hepatocyte proliferation in NAFLD-related cirrhosis. EGFR activation can induce lipogenic gene expression, contributing to NAFLD; however, EGFR overexpression suppresses lipogenic genes such as SREBF1, FASN, ACC1, and PPARα via TGF-β signaling150. Heparin-binding EGF-like growth factor is implicated in insulin resistance development induced by oxidative stressors like endothelin-1, thrombin, and serotonin in adipocytes and skeletal muscle. Adiponectin sequesters heparin-binding EGF-like growth factor, potentially explaining its anti-atherogenic and anti-inflammatory properties151,152.
Heparin-binding EGF-like growth factor expressed and released from liver sinusoidal endothelial cells may contribute to dyslipidemia and low-grade hepatic inflammation. Saturated liver sinusoidal endothelial cells can induce extrahepatic endothelial activation, promoting systemic inflammation. Heparin-binding EGF-like growth factor transcription is upregulated by oxidative stress in endothelial cells, linking oxidative stress to low-grade inflammation in obese NAFLD patients153. Endothelial-specific Notch activation, observed in capillarized liver sinusoidal endothelial cells in NAFLD/NASH, reduces secretion of hepatocyte mitogens including Wnt2a, Wnt9b, and HGF154.
Non-invasive biomarkers reflect distinct metabolic signatures in NAFLD, including hepatic HGF levels concurrent with N-palmitoyl-sphinganine155. Lipid metabolism enzymes influenced by NAFLD coincide with upregulated HGF and albumin, suggesting enhanced hepatocyte function and regenerative capacity156.
Activation of the HGF/c-Met pathway reduces transforming growth factor-β1 levels and serum alanine and aspartate aminotransferases, improving NASH liver function and inhibiting inflammation157,158. HGF administration prevents hepatic steatosis and ameliorates liver inflammation and fibrosis in rat models, highlighting its hepatoprotective role in NASH pathogenesis159. Despite these insights, the precise modulation of HGF function during NAFLD onset remains underexplored.
Validation of growth factors and pancreatic hormones as reliable biomarkers of NAFLD
Variability in sampling and inconsistencies in interpreting liver tissue examinations160,161 have driven efforts to identify alternative biomarkers162,163 and imaging tools for evaluating hepatic injury, particularly fibrosis163,164. Despite these challenges, liver biopsy remains the gold standard for assessing hepatic damage165, with confidence strengthened by standardized histological protocols166. However, validated biomarkers for predicting disease risk and therapeutic response in NAFLD remain lacking and are urgently needed.
Given the high prevalence of low-risk MASLD, it is crucial to detect significant fibrosis in individuals with high-risk features such as type 2 diabetes, abdominal obesity, or multiple cardiometabolic risk factors. First-line strategies include further stratification using second-line tests like transient elastography, followed by evaluation of whether patients with MASLD are receiving adequate management of cardiometabolic comorbidities. Non-invasive methods are increasingly central for fibrosis staging and disease monitoring. These tools can be grouped into serum biomarkers and scoring systems—effective for ruling out advanced fibrosis—and imaging techniques that measure liver stiffness, which better predict advanced fibrosis167. Such approaches are also useful for monitoring fibrosis progression, estimating survival, and predicting liver-related outcomes168.
Although biopsy remains necessary in selected cases to exclude concurrent liver conditions, non-invasive methods are progressively replacing it for fibrosis evaluation.
While biopsy remains the standard in clinical research, its limitations include invasiveness and sampling variability. Frequently used blood biomarkers include alanine and aspartate aminotransferases, low-density lipoprotein, high-density lipoprotein, and TG, often elevated in hyperlipidemia. Imaging techniques include ultrasound, fibroscan, and magnetic resonance modalities. Given the limitations of each, steatosis and fibrosis assessments should integrate serological and imaging methods. The Fibrosis-4 index, based on age, alanine and aspartate aminotransferases, and platelet count, is widely applied for prognosis, screening, and stratification.
Considering the inflammatory nature of NAFLD, biomarkers such as IL-1β, IL-6, IL-8, IL-10, and tumor necrosis factor-α are significantly elevated in NAFLD/NASH patients compared with healthy controls and those with simple steatosis, with tumor necrosis factor-α notably higher in simple steatosis relative to controls169.
Several studies have identified potential biomarker panels, including a set of 12 molecules measured by High-Sensitivity Cytokine Array I: EGF, interferon-γ, IL-1α, IL-1β, IL-2, IL-4, IL-6, IL-8, IL-10, monocyte chemoattractant protein-1, tumor necrosis factor-α, and vascular endothelial growth factor170. Other combinations investigated include FABP-1, PIIINP, ST2/IL-33R, albumin, alanine and aspartate aminotransferases. From these, three inflammatory markers (IL-6, IL-8, tumor necrosis factor-α) and two fibrosis markers (PIIINP, ST2/IL-33R) emerged as promising candidates for validation. To evaluate hepatic fibrosis—including steatohepatitis and cirrhosis—non-invasive scores, vibration-controlled transient elastography, imaging, and both direct and indirect serum biomarkers are applied alongside biopsy171. Direct markers reflect extracellular matrix remodeling and fibrosis172.
Combining serologic assays with transient elastography enhances sensitivity and specificity173. Prognostic scores such as FIB-4, the NAFLD Fibrosis Score, and the aspartate aminotransferase-to-Platelet Ratio Index (APRI) remain standard for predicting outcomes and mortality.
Research into molecular mechanisms of NAFLD has revealed new biomarkers and therapeutic targets174. Among these are circulating cell-free DNA and their methylation profiles175, as well as microRNAs, short non-coding RNAs with key regulatory roles in hepatic disease. MASLD alters hepatic miRNA expression at multiple disease stages, with several species implicated in progression from steatosis to NASH and cirrhosis176. Circulating microRNAs such as microRNA-34a, microRNA-122, and microRNA-192 are promising for diagnosis and staging. For instance, one study showed that these miRNAs reliably distinguish MASLD from healthy controls, with microRNA-34a further separating NASH from NAFLD177. Although further studies are needed, circulating microRNAs represent strong biomarker candidates. On the note, microRNA-22 modulates multiple pathways and epigenetic processes, functioning as both a metabolic regulator and a tumor suppressor in hepatocellular carcinoma174.
Hepatokines – including Fetuin-A, Fetuin-B, fibroblast growth factor 21, retinol-binding protein 4, angiopoietin-like protein 8, leukocyte cell-derived chemotaxin 2, and selenoprotein P play critical roles in hepatic lipid metabolism, inflammation, and systemic insulin resistance178. Among these, Fetuin-A, fibroblast growth factor 21, and angiopoietin-like protein 8 are especially promising for early diagnosis and therapeutic applications, though evidence is still exploratory. Hepatokines thus represent compelling biomarker and therapeutic candidates, reflecting the complexity of the liver–adipose–muscle axis. Yet, variability in study design, assay methods, and disease phenotypes hampers clinical translation, emphasizing the need for systematic reviews and large-scale standardized studies179. Effective clinical implementation will require hepatokine panels integrated with imaging and metabolic assessments.
Mechanistically, Fetuin-A inhibits insulin receptor tyrosine kinase activity, promoting ectopic lipid deposition and pro-inflammatory cytokine release180. Elevated Fetuin-A levels in lean NAFLD patients challenge the view of NAFLD as an obesity-driven condition, instead pointing to broader metabolic dysfunction. Angiopoietin-like protein 8, secreted in response to feeding and insulin, may exacerbate hepatic steatosis by modulating lipoprotein lipase and triglyceride clearance181. Fibroblast growth factor 21, a hepatokine with beneficial metabolic effects, is upregulated in NAFLD, likely as a compensatory response to lipid overload and oxidative stress182.
Beyond proteins and microRNAs, extracellular vesicle lipids also influence metabolic regulation. Zhu et al. identified four urinary extracellular vesicle lipids—free fatty acid (18:0), LPC (22:6/0:0), free fatty acid (18:1), and phosphatidyl inositol (16:0/18:1)—capable of distinguishing MASH from MAFL and reflecting fibrosis stage183.
As highlighted, NAFLD involves hepatocellular lipid accumulation (steatosis) associated with insulin resistance91 and glucagon resistance, leading to elevated glucagon and amino acid levels140,141. Neither circulating insulin nor glucagon are reliable NAFLD biomarkers. Using a high-sensitivity array in diverse liver diseases, seven biomarkers—EGF, interferon-γ, IL-1β, IL-6, IL-8, IL-10, and tumor necrosis factor-α—showed significant differences across groups, with no overlap between disease categories169. Serum EGF, for example, was significantly higher in hepatocellular carcinoma than in hepatitis C–related cirrhosis, supporting its use in hepatocellular carcinoma diagnosis, prognosis, and recurrence monitoring184. Likewise, HGF has shown value in identifying ectopic fat depots and predicting NAFLD onset. In a population-based cohort, HGF was the only biomarker consistently linked to steatotic liver disease and fibrosis onset by transient elastography185. These findings underscore HGF’s role in both HCV- and NAFLD-related conditions, correlating with disease stage and serving as a marker of hepatic fibrotic and inflammatory injury.
Recommendation for future research
The prevalence of MASLD continues to increase, particularly among individuals with obesity, underscoring the urgent need for early and accurate diagnosis, which is essential for effective detection and management. Advances in medical technology have accelerated the development and implementation of non-invasive tests, which enhance diagnostic accuracy while reducing reliance on liver biopsy. However, two critical factors—gender and age—should be carefully considered in the future design and application of NITs for NAFLD/MASH.
Recent studies have revealed gender-specific differences in the regulation of glucose, albumin, nitrogen-related metabolites, and hormones such as insulin and glucagon. For instance, serum insulin levels were found to be lower in men than in healthy women, whereas total bilirubin levels were higher in men, suggesting a role for sex hormones in these differences18. An analysis of the relationship between the Homeostatic Model Assessment for Insulin Resistance (HOMA-IR) and HGF in control groups showed no correlation in women, possibly reflecting a diminished association between insulin and HGF in this population. Among diabetic patients, gender-related differences were again evident: diabetic men displayed a weak negative correlation between glucose levels and HGF, whereas with EGF, weak correlations were detected in both genders of control subjects and in men with T2DM. In contrast, no correlation was observed in diabetic women18.
Notably, male subjects, whether control or diabetic, exhibited stronger associations with EGF, reaching moderate correlations, while female subjects showed a complete loss of correlation. These findings highlight significant gender-mediated differences in the signaling roles of HGF and EGF. They further emphasize the importance of insulin and glucagon as central regulators of glucose and lipid metabolism, particularly through their ratio, which appears to influence these growth factors18.
Moreover, fluctuations in serum glucose, albumin, bilirubin, ammonia, and nitric oxide (measured as nitrites) may contribute to variations in HGF and EGF, thereby affecting hepatic metabolic regulation. These alterations are especially pronounced in T2DM; a condition characterized by profound metabolic imbalance and impaired cellular proliferation, where gender and pathology significantly modulate the interplay between growth factors and liver metabolism18.
Taken together, these observations stress the necessity of incorporating gender-specific differences into future efforts to validate and refine biomarkers for NAFLD. Such consideration will be crucial for ensuring the reproducibility, specificity, and clinical utility of novel non-invasive tests.
Conclusion
NAFLD has emerged as one of the most prevalent causes of chronic liver disease, and its global incidence continues to rise. Early therapeutic intervention could mitigate progression and reduce the overall burden of liver disease. Achieving this goal requires the development of increasingly specific biomarkers derived from non-invasive tests to aid in the diagnosis and management of complications associated with NAFLD.
NAFLD is pathologically characterized by the accumulation of lipid droplets within hepatocytes (steatosis), hepatocyte stress, lipid peroxidation, and inflammation, which can progress to non-alcoholic steatohepatitis (NASH), metabolic dysfunction-associated steatohepatitis, hepatic fibrosis, and cirrhosis. In this disease context, pancreatic hormones (insulin and glucagon) and growth factors (HGF and EGF) are central to hepatic metabolism and physiology, processes that are profoundly disrupted in NAFLD. Consequently, these hormones and growth factors are increasingly being evaluated as candidate biomarkers, among other molecules with diagnostic potential. Although both insulin resistance and the recently described glucagon resistance play major roles in the pathogenesis of NAFLD, serum concentrations of these hormones do not strongly correlate with disease onset. By contrast, growth factors show greater promise: EGF has been identified as a reliable marker for the presence of hepatocellular carcinoma and its metastatic spread, whereas HGF may serve as a marker of chronic pathological processes such as steatosis and fibrosis. However, additional studies are needed to fully validate its clinical relevance.
In conclusion, integrating pancreatic hormones and growth factors into biomarker research offers significant potential to improve diagnostic precision and disease stratification. Future studies should account for age- and gender-specific influences to maximize the clinical applicability of NIT-based biomarkers in NAFLD and its related conditions.
Conflict of Interest Statement: None.
Funding Statement: The present investigation was partially funded by a grant # IN210623 from PAPIIT – DGAPA, Universidad Nacional Autónoma de México (UNAM).
References
- Powell EE, Wong VW, Rinella M. Non-alcoholic fatty liver disease. Lancet. 2021;397(10290):2212-2224.
- Muhamad NA, Maamor NH, Leman FN, et al. The global prevalence of nonalcoholic fatty liver disease and its association with cancers: Systematic review and meta-analysis. Interact J Med Res. 2023;12:e40653. doi: 10.2196/40653.
- Zahoor I, Mir GJ, Lone NA, Ashraf NU. Crosstalk between epigenetics and autophagy in metabolic dysfunction-associated steatotic liver disease. J Obes Metab Syndr. 2025;Jun 16. doi: 10.7570/jomes24041.
- Ashraf NU, Altaf M. Epigenetics: An emerging field in the pathogenesis of nonalcoholic fatty liver disease. Mutat Res Rev Mutat Res. 2018;778:1-12.
- Ahmed A, Wong RJ, Harrison SA. Nonalcoholic fatty liver disease review: Diagnosis, treatment, and outcomes. Clin Gastroenterol Hepatol. 2015;13(12):2062-2070.
- Rinella ME, Lazarus JV, Ratziu V, et al. NAFLD Nomenclature consensus group. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. J Hepatol. 2023;79(6):1542-1556.
- Dixon JB, Bhathal PS, O’Brien PE. Nonalcoholic fatty liver disease: predictors of nonalcoholic steatohepatitis and liver fibrosis in the severely obese. Gastroenterology. 2001;121(1):91-100.
- Rotter V, Nagaev I, Smith U. Interleukin-6 (IL-6) induces insulin resistance in 3T3-L1 adipocytes and is, like IL-8 and tumor necrosis factor-alpha, overexpressed in human fat cells from insulin-resistant subjects. J Biol Chem. 2003;278(46):45777-45784.
- Kuchay MS, Choudhary NS, Mishra SK. Pathophysiological mechanisms underlying MAFLD. Diabetes Metab Syndr. 2020;14(6):1875-1887.
- Sun Y, Hu D, Yu M, et al. Diagnostic accuracy of non-Invasive diagnostic tests for nonalcoholic fatty liver disease: A systematic review and network meta-analysis. Clin Epidemiol. 2025;17:53-71.
- Man S, Deng Y, Ma Y, et al. Prevalence of liver steatosis and fibrosis in the general population and various high-risk populations: A nationwide study with 5.7 million adults in China. Gastroenterology. 2023;165(4):1025-1040.
- Wang JL, Jiang SW, Hu AR, et al. Non-invasive diagnosis of non-alcoholic fatty liver disease: Current status and future perspective. Heliyon. 2024;10(5):e27325. doi: 10.1016/j.heliyon.2024.e27325.
- Bottaro DP, Rubin JS, Faletto DL, et al. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science. 1991;251(4995):802-804.
- Marquardt JU, Seo D, Gómez-Quiroz LE, et al. Loss of c-Met accelerates development of liver fibrosis in response to CCl(4) exposure through deregulation of multiple molecular pathways. Biochim Biophys Acta. 2012;1822(6):942-951.
- Fafalios A, Ma J, Tan X, et al. A hepatocyte growth factor receptor (Met)-insulin receptor hybrid governs hepatic glucose metabolism. Nat Med. 2011;17(12):1577-1584.
- Akhtar S, Benter IF. The role of epidermal growth factor receptor in diabetes-induced cardiac dysfunction. Bioimpacts. 2013;3(1):5-9.
- Song I, Patel O, Himpe E, Muller CJ, Bouwens L. Beta cell mass restoration in alloxan-diabetic mice treated with EGF and gastrin. PLoS One. 2015;10(10):e0140148. doi: 10.1371/journal.pone.0140148.
- Contreras-Zentella ML, Alatriste-Contreras MG, Suárez-Cuenca JA, Hernández-Muñoz R. Gender effect of glucose, insulin/glucagon ratio, lipids, and nitrogen-metabolites on serum HGF and EGF levels in patients with diabetes type 2. Front Mol Biosci. 2024;11:1362305. doi: 10.3389/fmolb.2024.1362305.
- Nakamura T, Nawa K, Ichihara A. Partial purification and characterization of hepatocyte growth factor from serum of hepatectomized rats. Biochem Biophys Res Commun. 1984;122(3):1450-1459.
- Stolz DB, Mars WM, Petersen BE, Kim TH, Michalopoulos GK. Growth factor signal transduction immediately after two-thirds partial hepatectomy in the rat. Cancer Res. 1999;59(16):3954-3960.
- Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. 2010;11(12):834-848.
- Zhang Y, Xia M, Jin K, et al. Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol Cancer. 2018;17(1):45. doi: 10.1186/s12943-018-0796-y.
- Sakai K, Aoki S, Matsumoto K. Hepatocyte growth factor and Met in drug discovery. J Biochem. 2015;157(5):271-284.
- Zhao Y, Ye W, Wang YD, Chen WD. HGF/c-Met: A key promoter in liver regeneration. Front Pharmacol. 2022;13:808855. doi: 10.3389/fphar.2022.808855.
- Comoglio PM, Giordano S, Trusolino L. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat Rev Drug Discov. 2008;7(6):504-516.
- Galimi F, Bagnara GP, Bonsi L, et al. Hepatocyte growth factor induces proliferation and differentiation of multipotent and erythroid hemopoietic progenitors. J Cell Biol. 1994;127(6 Pt1):1743-1754.
- Gallo S, Sala V, Gatti S, Crepaldi T. Cellular and molecular mechanisms of HGF/Met in the cardiovascular system. Clin Sci (Lond). 2015;129(12):1173-1193.
- Cahill EF, Kennelly H, Carty F, Mahon BP, English K. Hepatocyte growth factor is required for mesenchymal stromal cell protection against bleomycin-Induced pulmonary fibrosis. Stem Cells Transl Med. 2016;5(10):1307-1318.
- Cooper CS, Park M, Blair DG, Tainsky MA, Huebner K, Vande Woude GF. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature. 1984;311(5981):29-33.
- Michalopoulos GK. Advances in liver regeneration. Expert Rev Gastroenterol Hepatol. 2014;8(8):897-907.
- Mohammed FF, Khokha R. Thinking outside the cell: proteases regulate hepatocyte division. Trends Cell Biol. 2005;15(10):555-563.
- Michalopoulos GK. Liver regeneration. J Cell Physiol. 2007;213(2):286-300.
- Yanagita K, Nagaike M, Ishibashi H, Niho Y, Matsumoto K, Nakamura T. Lung may have an endocrine function producing hepatocyte growth factor in response to injury of distal organs. Biochem Biophys Res Commun. 1992;182(2):802-809.
- Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121(7):977-990. doi: 10.1016/j.cell.2005.04.014.
- Ghanm SE, Shebl NA, El Sayed IET, Abdel-Bary HM, Saad BF, Othman Saad W. Direct relationship between interleukin-10 gene polymorphism and hepatocellular carcinoma complicated by direct acting antiviral treatment of hepatitis C virus. Asian Pac J Cancer Prev. 2021;22(10):3203-3210.
- El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology. 2012;142(6):1264-1273.e1.
- Lai L, Chen J, Wei X, et al. Transplantation of MSCs overexpressing HGF into a rat model of liver fibrosis. Mol Imaging Biol. 2016;18(1):43-51.
- Li Z, Mizuno S, Nakamura T. Antinecrotic and antiapoptotic effects of hepatocyte growth factor on cholestatic hepatitis in a mouse model of bile-obstructive diseases. Am J Physiol Gastrointest Liver Physiol. 2007;292(2):G639-646.
- Kosai K, Matsumoto K, Funakoshi H, Nakamura T. Hepatocyte growth factor prevents endotoxin-induced lethal hepatic failure in mice. Hepatology. 1999;30(1):151-159.
- Yuge K, Takahashi T, Nagano S, et al. Adenoviral gene transduction of hepatocyte growth factor elicits inhibitory effects for hepatoma. Int J Oncol. 2005;27(1):77-85.
- Hardesty JE, Wahlang B, Prough RA, Head KZ, Wilkey D, Merchant M, Shi H, Jin J, Cave MC. Effect of epidermal growth factor treatment and polychlorinated biphenyl exposure in a dietary-exposure mouse model of steatohepatitis. Environ Health Perspect. 2021;129(3):37010. doi: 10.1289/EHP8222.
- Xu H, Liu L, Cong M, Liu T, Sun S, Ma H. EGF neutralization antibodies attenuate liver fibrosis by inhibiting myofibroblast proliferation in bile duct ligation mice. Histochem Cell Biol. 2020;154(1):107-116.
- Modica TME, Dituri F, Mancarella S, Pisano C, Fabregat I, Giannelli G. Calcium regulates HCC proliferation as well as EGFR recycling/degradation and could be a new therapeutic target in HCC. Cancers (Basel). 2019;11(10):1588. doi: 10.3390/cancers11101588.
- Cao S, Pan Y, Tang J, Terker AS, Arroyo Ornelas JP, et al. EGFR-mediated activation of adipose tissue macrophages promotes obesity and insulin resistance. Nat Commun. 2022;13(1):4684. doi: 10.1038/s41467-022-32348-3.
- Orofiamma LA, Vural D, Antonescu CN. Control of cell metabolism by the epidermal growth factor receptor. Biochim Biophys Acta Mol Cell Res. 2022;1869(12):119359. doi: 10.1016/j.bbamcr.2022.119359.
- Aydın MM, Akçalı KC. Liver fibrosis. Turk J Gastroenterol. 2018;29(1):14-21.
- Shehata F, Abdel Monem N, Sakr M, Kasem S, Balbaa M. Epidermal growth factor, its receptor and transforming growth factor-β1 in the diagnosis of HCV-induced hepatocellular carcinoma. Med Oncol. 2013;30(3):673. doi: 10.1007/s12032-013-0673-x.
- Bosch F, Bouscarel B, Slaton J, Blackmore PF, Exton JH. Epidermal growth factor mimics insulin effects in rat hepatocytes. Biochem J. 1986;239(3):523-530.
- Soler C, Galan X, Peinado-Onsurbe J, Quintana I, Llobera M, Ramírez I. Epidermal growth factor interferes with the effect of adrenaline on glucose production and on hepatic lipase secretion in rat hepatocytes. Regul Pept. 1993;44(1):11-16.
- Peak M, Agius L. Inhibition of glycogen synthesis by epidermal growth factor in hepatocytes. The role of cell density and pertussis toxin-sensitive GTP-binding proteins. Eur J Biochem. 1994;221(1):529-536.
- Abu Rmilah AA, Zhou W, Nyberg SL. Hormonal contribution to liver regeneration. Mayo Clin Proc Innov Qual Outcomes. 2020;4(3):315-338.
- Kimura M, Moteki H, Ogihara M. Role of hepatocyte growth regulators in liver regeneration. Cells. 2023;12(2):208. doi: 10.3390/cells12020208.
- Tokarz VL, MacDonald PE, Klip A. The cell biology of systemic insulin function. J Cell Biol. 2018;217(7):2273-2289.
- Ohlsson B, Jansen C, Ihse I, Axelson J. Epidermal growth factor induces cell proliferation in mouse pancreas and salivary glands. Pancreas. 1997;14(1):94-98.
- Mehta DI, Horváth K, Chanasongcram S, Hill ID, Panigrahi P. Epidermal growth factor up-regulates sodium-glucose cotransport in enterocyte models in the presence of cholera toxin. JPEN J Parenter Enteral Nutr. 1997;21(4):185-191.
- Schneider JA, Diamond I, Rozengurt E. Glycolysis of quiescent cultures of 3T3 cells. Addition of serum, epidermal growth factor, and insulin increases the activity of phosphofructokinase in a protein synthesis-independent manner. J Biol Chem. 1978;253(3):872-877.
- Wang Z. Transactivation of epidermal growth factor receptor by G Protein-Coupled Receptors: Recent progress, challenges and future research. Int J Mol Sci. 2016;17(1):95. doi: 10.3390/ijms17010095.
- López-Luque J, Caballero-Díaz D, Martinez-Palacián A, et al. Dissecting the role of epidermal growth factor receptor catalytic activity during liver regeneration and hepatocarcinogenesis. Hepatology. 2016;63(2):604-619.
- Chen J, Zeng F, Forrester SJ, Eguchi S, Zhang MZ, Harris RC. Expression and function of the epidermal growth factor receptor in physiology and disease. Physiol Rev. 2016;96(3):1025-1069.
- Cao S, Pan Y, Tang J, Terker AS, Arroyo Ornelas JP, et al. EGFR-mediated activation of adipose tissue macrophages promotes obesity and insulin resistance. Nat Commun. 2022;13(1):4684. doi: 10.1038/s41467-022-32348-3.
- Orofiamma LA, Vural D, Antonescu CN. Control of cell metabolism by the epidermal growth factor receptor. Biochim Biophys Acta Mol Cell Res. 2022;1869(12):119359. doi: 10.1016/j.bbamcr.2022.119359.
- Aydın MM, Akçalı KC. Liver fibrosis. Turk J Gastroenterol. 2018;29(1):14-21.
- Shehata F, Abdel Monem N, Sakr M, Kasem S, Balbaa M. Epidermal growth factor, its receptor and transforming growth factor-β1 in the diagnosis of HCV-induced hepatocellular carcinoma. Med Oncol.