





Sickle Cell Disease in Angola: Public Health Challenges
Sickle Cell Disease in Angola as a Public Health Issue
Francisco João Pinto*, Angola. Anemia falciforme. 1, Scott D Grosse 2, Isaac Odame 2, Hani K Atrash 2, Djesika D Amendah 2, Frédéric B Piel 2, Thomas N Williams 2, Patrick T McGann 3, Margaret G Ferris 3, Uma Ramamurthy 3, Brigida Santos 3, Vysolela de Oliveira 3, Luis Bernardino 3, Russell E Ware 3,. Berenice Assumpção Kikuchi 4, Maria Lucia Ivo 5, Rita de Cassia Mousinho-Ribeiro 6, Greice Lemos Cardoso 7, eItallo Esteves Lacerda de Sousa 8 , Person Priscila K.C:Martins 8. Prithu Sundd 9, Mark T Gladwin 9, Enrico M Novelli 9. Madelyn S Gould 10, Frank A Marrocco10. Marjorie Kleinman10, John Graham Thomas10. Katherine Mostkoff10. Jean Cote10. Mark Davies10. D Creel11, C J Witkop Jr12, R A King13. Amanda Simons14. Rebat M. Halder M.D.15, Sharon Bridgeman-Shah M.D16. C. Kruijt17. L. Montoliu18. Viktoria C Brücher 19, Peter Heiduschka 19, Ulrike Grenzebach 19, Nicole Eter 19, Julia Biermann 19. Hoffmann MB20 .M. Vittoria Schiaffino21, Carlo Tacchetti22. G. Keren23. C. Lewis23. Marjan Huizing 24, May C V Malicdan 24, Jennifer A Wang 24, Hadass Pri-Chen 25, Richard A Hess 24, Roxanne Fischer 24, Kevin J O’Brien 26, Melissa A Merideth 26, William A Gahl 24, Bernadette R Gochuico 24. G. Chiang 27, Wenqing lu27, Xiangshu Piao27, J.K. Hu27. Mariangela Mancini28, Sophie Camilleri-Bröet28, Benjamin O. Anderson29, David M. Hockenbery30. CRA.Constituição da Rep.ublica de Angola31. Henry W Lim 32, Indermeet Kohli 33, Eduardo Ruvolo 34, Ludger Kolbe 34, Iltefat H Hamzavi 33. D Lvovs 35, O O Favorova35, A V Favorov35. David Bick 36, Sarah L Bick 37, David P Dimmock 38, Tom A Fowler39, Mark J Caulfield 40, Richard H Scott 41.Tara Rodden Robinson42. Edward Willett43. Priscila Neves Costa44. MINSA. Ministério da Saúde de Angola. Anemia Falceforme45. MINSA. População Angolana com traço falciforme46. David C Rees 47, Thomas N Williams47. Mark T Gladwin47. K J Wierenga 48, I R Hambleton48, N A Lewis48. Vinay Kumar MBBS MD FRCPath49 , Abul K. Abbas MBBS50 , Jon C. Aster MD PhD51. Obi Peter Adigwe 52, Solomon Oloche Onoja 53, Godspower Onavbavba 52. E. S. Azevedo54. A. Y. Elzouki 55, H. A. Harfi, H. Nazer55, William Oh , F. B55. Stapleton , R. J. Whitley55 .K. J. Wierenga56, I. R. Hambleton 56 .N. A. Lewis56. KC Aggarwal57. PC Goyal Prasad57. S Saluja,M Sharma57. Neeraj Awasthy57
- Agostinho Neto University. UNINET – Center for Studies, Scientific Research and AdvancedTraining in Computer Systems andCommunication University Campus of theComama, S/N, LuandaAngola Correspondingauthor:[email protected] Republica de Angola.
- Division of Blood Disorders, National Center on Birth Defects and Developmental Disabilities, CDC, Atlanta, Georgia 30333, USA.
- Department of Pediatrics, Baylor College of Medicine, Houston, Texas. Associação de Anemia Falciforme do estado de São-Brasil
- Universidade Federal do Mato Grosso do Sul-Brasil.
- Universidade Federal do Pará, Instituto de Ciências Biológicas, Pará, Brasil.
- School Universidade Federal do Pará, Programa de Pós-Graduação em Genética eBiologia, Pará, Brasil.
- School Centro Universitário do Pará, Pará, Belém, Brasil.
- Sickle Ce ll Center of Excellence, University of Pittsburgh School of Medicine, Pittsburgh,Pennsylvania 15261, USA.
- Division of Child and Adolescent Psychiatry, Columbia University and New York State Psychiatric Institute, New York, NY 10032, USA.
- Phoenix Veterans Administration Hospital and Arizona State University, University of Minnesota Dental and Medical Schools, Minneapolis, Minn.
- Division of Human and Oral Genetics, Phoenix, Ariz., University of Minnesota Dental and Medical Schools, Minneapolis, Minn.
- Department of Medicine, University of Minnesota Dental and Medical Schools, Minneapolis, Minn.
- Framingham State University, through the Remixing Open Textbooks with an Equity Lens (ROTEL) project.
- Department of Dermatology, Howard University College of Medicine, Washington, DC.
- Department of Dermatology, Howard University Hospital, 2041 Georgia Avenue NW, Washington, DC 20060.
- Department of Human Genetics, Amsterdam UMC, and the Netherlands Institute for Neuroscience.
- Department of Molecular and Cellular Biology, National Centre for Biotechnology (CNB-CSIC), CIBERER-ISCIII, Madrid, Spain.
- Dept. of Ophthalmology, University of Muenster Medical Centre, Muenster, Germany.
- Visual Processing Lab – Dept Ophthalmology – Otto-von-Guericke-University Magdeburg.
- IBIT, Scientific Institute San Raffaele, Via Olgettina 58, 20132 Milan, Italy.
- Department of Experimental Medicine, University of Genoa Medical School, Via De Toni 14, 16132 Genoa, Italy.
- Lawrence Erlbaum Associates, Inc. (in Hillsdale, NJ) is the publisher.
- Human Biochemical Genetics Section, Medical Genetics Branch, National Human Genome Research Institute, National Institutes of Health, Bethesda, Maryland.
- Molecular Disease Unit, Edmond and Lily Safra Children’s Hospital, Sheba Medical Center, Tel Hashomer and Sackler Faculty of Medicine, Tel Aviv University, Tel Aviv, Israel.
- Office of the Clinical Director, National Human Genome Research Institute of Health, Bethesda, Maryland.
- Thacker College of Animal Science and Technology, China Agricultural University, No. 2 Yuanmingyuan West Road, Beijing, Post Code 100193, China.
- University of Padua, Padova, Italy.
- Professor Emeritus at University of Washington.
- Faculty Member at Fred Hutch Cancer Center.
- Republica de Angola. Orgão Oficial I Série, Nº 154 de 16 de Agosto de 2023.
- Photomedicine and Photobiology Unit, Department of Dermatology,
- Henry Ford Health, Detroit, Michigan.
- Photomedicine and Photobiology Unit, Department of Dermatology,Henry Ford Health, Detroit, Michigan.
- Beiersdorf, Inc., Morristown, New Jersey.
- Scientific Center of Russian Federation Research Institute for Genetics and Selection of Industrial Microorganisms “Genetika”, 1-st Dorozhny proezd, 1, Moscow, Russia, 113545.
- HudsonAlpha Institute for Biotechnology, Huntsville, Alabama, USA.
- Nemours/Alfred I. duPont Hospital for Children, Wilmington, Delaware, USA.
- Rady Children’s Institute for Genomic Medicine, San Diego, California, USA.
- Genomics England Ltd., London, UK.
- William Harvey Research Institute, Queen Mary University of London, London, UK.
- Department of Clinical Genetics, Great Ormond Street Hospital for Children,National Health Service (NHS) Foundation Trust, London, UK.
- Genetics Dummies Oregon State University.
- Genetics Demystified. New York: McGraw-Hill. 2006.
- Faculdade de Educação e Tecnologia da Amazônia, PA, Brasil
- Republica de Angola.
- Republica de Angola.
- Department of Paediatric Haematology, King’s College Hospital NHS Foundation Trust, King’s College London
- Sickle Cell Unit, Tropical Medicine Research Institute, University of the West Indies, Mona, Kingston, Jamaica.
- Department of Pathology, University of Chicago.
- Department of Pathology, University of California, San Francisco.
- Department of Pathology, University of Washington, school of medicine, Seattle, Washington.
- Office of the Director General, National Institute for Pharmaceutical Research and Development, Abuja, Federal Capital Territory, Nigeria.
- Department of Medical Laboratory Sciences, University of Nigeria, Enugu, Nigeria.
- Gazeta Médica da Bahia, da Universidade Federal da Bahia (UFBA) – Brasil.
- Textbook of Clinical Pediatrics 2ª ed. 2012 Edição. Springer, England.
- Survival estimates for patients with homozygous sickle-cell disease in Jamaica: A clinic-based population study.
- Tertiary care center in north India.
OPEN ACCESS
PUBLISHED: 30 November 2025
CITATION: Pinto, F.J., Falciforme, AA., et al., 2025. Sickle Cell Disease in Angola as a Public Health Issue. Medical Research Archives, [online] 13(11). https://doi.org/10.18103/mra.v13i11.6997
COPYRIGHT: © 2025 European Society of Medicine. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
DOI: https://doi.org/10.18103/mra.v13i11.6997
ISSN 2375-1924
ABSTRACT
In this paper, we describe sickle cell disease as a public health issue in Angola. Angola has an estimated population of around 34 million, with approximately 18% to 20% of people affected. In 2022, 924 new cases were recorded, and studies indicate that the disease is one of the leading causes of infant death in the country, even with some progress in infection control. Studies also indicate that, in the same year, approximately 1,000 children died, and it is estimated that 1,000 children die annually and more than 7,000 children are born with the disease each year. It is a disease that causes deformation of red blood cells, leading to symptoms such as chronic anemia, intense pain due to obstruction of blood vessels, and increased susceptibility to infections, which results in suffering and premature death. In sickle cell disease, the HbSS genotype of sickle cell anemia, considered the most severe form, prevalent in the African continent, has high morbidity and mortality. In this work, we used quantitative observation as a research method, which allowed us to collect numerical data about the phenomenon. It also allowed us to quantify the problem and understand its dimension, as well as to investigate and observe the phenomenon comprehensively.
Keywords:
- Sickle cell disease
- Angola
- Public health issue
1 Introduction
The general objective of this article is to describe sickle cell disease as a public health issue in Angola. Sickle cell anemia is an inherited disease that affects a significant portion of the Angolan population and it is one of the main public health problems in the country, especially among children. It is characterized by the deformation of red blood cells, leading to serious complications such as pain crises, chronic anemia, and organ damage, with high rates of morbidity and mortality. The diagnosis and implementation of a National Comprehensive Care Program are crucial for the treatment of the disease in Angola.
Sickle cell anemia is a serious and common hereditary disease in Angola, affecting up to 20% of the population and causing around a 1,000 new cases per year. The condition is caused by the deformation of red blood cells into a sickle shape, which leads to obstruction of blood vessels, causing pain attacks, chronic anemia and a predisposition to infections. There is an ongoing effort to improve care for people with the disease, including the implementation of national policies and the training of health professionals.
1.1 HISTORY OF SICKLE CELL ANEMIA IN ANGOLA
The history of sickle cell anemia in Angola is linked to its high prevalence, with estimates suggesting that around 20% of the Angolan population is affected by this hereditary disease. Although the disease has been known for centuries in Africa, recent studies in Angola have revealed an increase in new cases, with approximately 1,000 to 7,000 children born with the disease annually, and more than 1,000 deaths recorded between 2011 and 2020. The lack of government subsidies for treatment and widespread ignorance about the disease, including diagnostic tests, are significant challenges.
1.2 SICKLE CELL DISEASE GROUP
The term sickle cell disease refers to a heterogeneous group of clinical conditions with a genetic basis, whose genotype includes at least one allele that encodes the mutated form of hemoglobin – hemoglobin S (HbS). The sickle cell disease group includes several genetic conditions that share the presence of hemoglobin S (HbS), which can deform red blood cells into a sickle shape, hindering blood circulation. The most common forms are sickle cell anemia (HbSS), hemoglobin SC disease (HbSC), and hemoglobin S-beta-thalassemia (HbS/thalassemia). These diseases are inherited and can cause similar symptoms such as anemia, chronic pain, and infections, although the severity varies between types.
1.2.1 Main types of sickle cell disease
- Sickle cell anemia (HbSS): The most severe form, in which a person inherits two copies of the hemoglobin S gene (one from each parent).
- Hemoglobin SC (HbSC) disease: This occurs when a person inherits the hemoglobin S gene from one parent and a hemoglobin C gene from another. It is usually milder than sickle cell anemia.
- Hemoglobin S-beta-thalassemia (HbS/thalassemia): This results from inheritance of the hemoglobin S gene from one parent and the beta-thalassemia gene from the other. The severity depends on the type of thalassemia inherited, with HbS/beta-thalassemia-zero generally being more severe.
- Other forms: There are other rarer forms, such as those involving hemoglobin D (HbSD) or E (HbSE), which also fall into the sickle cell disease group.
Among sickle cell diseases, sickle cell anemia (HbSS) is the most serious and neglected socio-environmental disease due to the lack of effective public policies in Angola. Therefore, it is a public health issue. Mortality in Angola ranges from 50% to 90% in children up to 5 years of age. The scarcity of published scientific research on sickle cell disease makes it difficult to understand the social, economic and political context that articulates public policies. One published initiative that we are aware of was the prospective pilot program for neonatal screening and treatment for sickle cell anemia in Luanda, Angola.
The term sickle cell disease is attributed to a group of diseases, which from a genotypic point of view, corresponds to hemoglobinopathies, characterized by the presence of Hemoglobin S, in homozygosity and/or heterozygosity. Clinically manifested in three main syndromes: sickle cell anemia, S-β-thalassemia and hemoglobinopathy SC. Sickle cell anemia occurs due to a mutation in the beta globin gene, which results in the exchange of two amino acids: valine for glutamic acid at position six of the beta globin chain. This substitution leads to the polymerization of hemoglobin S under low oxygenation conditions, altering the rheology and survival of the erythrocyte. A disease that causes a high level of suffering to the person and their family, generally from the sixth month of life onwards due to the drop in HbF levels. The stress of living with the imminent threat of premature death permeates the daily lives of these people. Vasoocclusion crises due to red blood cell sickling cause pain in different parts of the body, increased susceptibility to recurrent infections, chronic anemia, and progressive involvement of several organs, high morbidity and early mortality. Although we mention some important aspects related to the topic throughout the work, in this study, the fundamental focus is to describe sickle cell anemia as a public health issue in Angola.
2 Theoretical foundation
Genetics is the study of genes and tries to explain what they are and how they work. Genes are living organisms inherit characteristics or traits from their ancestors; for example, children usually look like their parents because they have inherit their parents’ genes. Genetics tries to identify which characteristics are inherited and to explain how these characteristics are passed from generation to generation.
Some characteristics are part of an organism’s physical appearance, such as eye color or height. Other types of characteristics are not easily seen and include blood types. Some characteristics are inherited through genes, which is the reason why tall and thin people tend to have tall and thin children. Other characteristics come from interactions between genes and the environment. The way our genes and environment interact to produce a trait can be complicated. For example, the probability of somebody dying of cancer or heart disease seems to depend on both their genes and their lifestyle.
Genes are made from a long molecule called DNA (deoxyribonucleic acid), which is copied and inherited across generations. DNA is a molecule that contains the genetic information for an organism´s development and function. It is passed from one generation to the next. A mutation is a sequential change in DNA, which can occur naturally or be caused by external factors. This appearance of new characteristics is important in revolutionizing organism.
In biology, a mutation is an alteration in the nucleic acid sequence of the genome of an organism, or extra chromosomal DNA. It creates slightly different versions of the same genes, called alleles. These small differences DNA sequence make every individual unique.
2.1 GENES AND INHERITANCE
All organisms inherit the genetic information specifying their structure and function form their parents. Likewise, all cells arise from preexisting cells, so the genetic material must be replicated and passed from parent to progeny cell at each cell division. Genes are pieces of DNA that contain information for the synthesis of ribonucleic acids (RNAs) or polypeptides. They are inherited as units, with two parents dividing out copies of their genes to their offspring. Humans have two copies of each of their genes, but each egg or sperm cell only gets one of those copies for each gene. An egg and sperm join to form a zygote with a complete set of genes. The resulting offspring has the same number of genes as their parents, but for any gene, one of their two copies comes from their father and one from their mother.
Example of crossing: The effects of crossing depend on the types (the alleles) of the gene. If the father has two copies of an allele for red hair, and the mother has two copies for brown hair, all their children get the two alleles that give different instructions, one for red hair and one for brown. The hair color of these children depends on how these alleles work together. If one allele dominates the instructions from another, it is called the dominant allele, and the allele that is overridden is called the recessive allele. In the case of a daughter with alleles for both red and brown hair, brown is dominant and she ends up with brown hair.
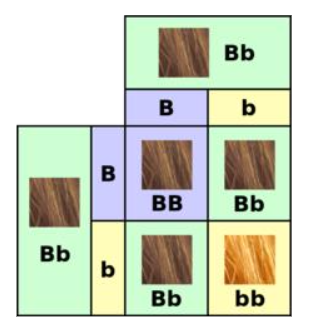
Now imagine that this woman and has children with a brown-haired man who also has a Bb genotype. Her ova will be a crossing of two types, one type containing the B allele, and one type the b allele. Similarly, her partner will produce a crossing of two types of sperm containing one or the other of these two alleles. When the transmitted genes are joined up in their offspring, these children have a chance of getting either brown or red hair, since they could get a genotype of BB = brown hair, Bb = brown hair or bb = red hair. In this generation, there is, therefore, a chance of the recessive allele showing itself in the phenotype of the children. Some of them may have red hair like their grandfather.
2.2 HOW GENES WORK
It consists of two major steps: transcription and translation. Together, transcription and translation are known as gene expression. During the process of transcription, the information stored in a gene’s DNA is passed to a similar molecule called RNA (ribonucleic acid) in the cell nucleus. The basic unit of heredity passed from parent to child. Genes are made up of sequences of DNA and are arranged, one after another, at specific locations on chromosomes in the nucleus of cells. The functional and physical unit of heredity passed from parent to offspring. Genes are pieces of DNA, and most genes contain the information for making a specific protein. Our genes carry information that gets passed from one generation to the next. For example, genes are why one child has blonde hair like their mother, while their sibling has brown hair like their father. Genes also determine why some illnesses run in families and whether babies will be male or female.
While the human genome contains an estimated 20,000 protein-coding genes, the coding segments of those genes—the exons—comprise less than 2% of the genome; most of the genome consists of DNA that lies between genes, far from genes or in vast areas spanning several million base pairs (Mb) that appear to contain no genes. The gene occurs in the same position on each chromosome. Genetic traits, such as eye color, are dominant or recessive: Dominant traits are controlled by 1 gene in the pair of chromosomes. Recessive characteristics need both genes in the gene pair to work together.
2.3 GENETIC DISORDER
A genetic disorder is a health problem caused by abnormalities in the genome. They are heritable, and may be passed down from the parents’ genes to their children and to later generations. If a genetic disorder is present from birth, it is described as a congenital defect. Some defects only show up in later life. The mutation responsible can occur spontaneously before the embryo develops, or it can be inherited from parents who are carriers of a faulty gene. There are well over 6,000 known genetic disorders, and new genetic disorders are constantly being found. More than 600 genetic disorders are treatable. Around in 50 people are affected by a known single-gene disorder, while around 1 in 263 are affected by a disorder caused by their chromosomes. Parts of a chromosome may be absent, or duplicated. About 65% of people have some kind of health problem as a result of congenital genetic mutations. About 1 in 21 people are affected by a genetic disorder classified as “rare” (less than 1 in 2,000 people). Most genetic disorders are rare in themselves. They may affect one person in every several thousands or even millions. Sometimes they are relatively frequent in a population. If they are frequent, it suggests these recessive gene disorders give an advantage in certain environments when only one copy of the gene is present. Sickle cell anemia is an example of this. The same disease, such as some forms of cancer, may be caused by an inherited genetic condition in some people, by new mutations in other people, and by non genetic causes in still other people. A disease is only called a genetic disease if it can be inherited at birth. The particular defect may only show up later in life.
3 Methodology
In this work, we used quantitative observation as a research method, which allowed us to collect numerical data about the phenomenon. It also allowed us to quantify the problem and understand its dimension, as well as to investigate and observe the phenomenon comprehensively. This is a quantitative observational study, which used Google Scholar and Virtual Health Library databases. The search strategy used the descriptors “Sickle cell disease”, “Angola” and “Public health issue”, 350 results were found, but after reading the titles, 10 articles were selected, according to the inclusion criteria of addressing the topic of sickle cell disease, which were read entirety. Furthermore, official documents from the Angolan Ministry of Health were analyzed, based on the Virtual Health Library database, and from the High Commissioner for Human Rights of the United Nations, in its annual report on a people with sickle cell disease in the world.
Quantitative observation as a research method was used to collect numerical data and measure specific variables systematically and objectively. It involves the observation and measurement of phenomena, events, behaviors, or characteristics to collect quantitative data for analysis and interpretation, and focuses on objectivity, accuracy, and quantification of relationships, making it a powerful approach for understanding and interpreting various phenomena in the world.
3.1 SICKLE CELL DISEASE IN ANGOLA
Sickle cell disease is a major issue in Angola due to its high prevalence, with estimates indicating that around 20% of Angolans may carry the sickle cell trait, and other data indicate that around 18% of the population has the trait, with around 1,000 children affected annually. It is a disease that causes deformation of red blood cells, leading to symptoms such as chronic anemia, severe pain due to obstruction of blood vessels, and increased susceptibility to infections, which results in suffering and premature deaths. Therefore, this is a serious public health issue in Angola.
3.1.1 Impact and challenges in Angola
In this subsection, we describe the impact and challenges of sickle cell disease in Angola:
- High prevalence: Angola has one of the highest incidences of the disease in Africa, affecting a large number of children and families.
- Lack of specialized care: There is a shortage of hematology specialists, especially in provinces like Malanje, which hinders patients’ access to adequate treatment.
- Lack of state subsidy: Currently, the disease does not receive state subsidies, which impedes access to treatment and tests (such as hemoglobin electrophoresis), especially in the private healthcare sector.
- International support: Angola requests support from Brazil and other nations to develop a National Comprehensive Care Program for people with sickle cell disease.
- Professional training: There are projects to train healthcare professionals in topics such as self-care, neonatal screening, and comprehensive quality care.
- Public awareness: Awareness campaigns are needed to promote voluntary blood donation and awareness of the disease.
3.1.2 Symptoms and Severity
- Pain: Red blood cell sickling causes pain crises in various parts of the body, known as vaso-occlusion.
- Chronic Anemia: Patients suffer from anemia, which causes unexplained fatigue.
- Complications: The disease can lead to damage to various organs and increases susceptibility to injections.
- Unexplained fatigue
- Swelling, redness, and pain in the hands and feet
- Joint or abdominal pain
- Jaundice (yellowing of the eyes and skin) Infections
3.1.3 Causes of sickle cell disease
- Genetic Mutation: Sickle cell disease is caused by a mutation in the gene that produces hemoglobin, the protein inside red blood cells that carries oxygen.
- Genetic Inheritance: The condition is passed from parents to children, and both parents must carry the mutation for a child to have the disease.
3.1.4 Diagnosis
The heel prick test, performed in the baby’s first week of life, can detect sickle cell anemia. It is done through blood tests, such as hemoglobin electrophoresis, to identify the presence of mutant hemoglobin.
3.1.5 Treatment
There is no cure, but treatment and specialized monitoring can improve quality of life and life expectancy. Treatments help control symptoms and prevent complications. This includes:
- Preventative antibiotics: To help prevent infections in children.
- Hydroxyurea: Medications that can help reduce the frequency of pain attacks.
- Blood transfusions: May be used in some cases.
- Folic acid supplementation: A vitamin that helps in the production of red blood cells.
3.1.6 Care
- Live a healthy lifestyle, without excesses
- Eat a diet rich in vegetables, fruits, and meat
- Drink plenty of water
- Wear light, cool clothing
- Wear socks and shoes that don’t hurt your feet
- Drink more water and take pain medication in case of mild pain attacks without fever
- Go to the hospital if you have a fever or pain that doesn’t go away
3.1.7 Prevention
- Promote the training of healthcare professionals
- Improve and expand neonatal screening
- Raise awareness about voluntary blood donation
4 Results and Discussion
In this section we present the results obtained and their discussion.
4.1 RESULTS
In Angola, sickle cell anemia affects about 20% of the population and is linked to high infant mortality rates. In 2022, 924 new cases were recorded in children, and studies indicate that more than 1,000 children died from the disease between 2011 and 2020. The Ministry of Health estimates that between 60,000 and 100,000 Angolans have the disease, with more than 3 million carriers of the sickle cell trait. Sickle cell disease is genetic in origin and its management faces infrastructure challenges and the lack of state subsidies for treatment. The disease causes chronic anemia and pain crises. This disease is a public health challenge in Angola, affecting a significant portion of the population. Estimates indicate that approximately 20% of Angolans may carry the sickle cell trait, and approximately 1,000 children are born with the disease annually. International cooperation efforts, such as the partnership with Brazil, aim to create national policies, strengthen neonatal screening, and train health professionals to better care for patients across the country.
4.1.1 Impact and estimation
- Prevalence: It is estimated that approximately 20% of the Angolan population carries the sickle cell trait, with more than 3 million people living with the disease.
- Mortality: The disease is one of the leading causes of infant mortality, with more than 1,000 children dying annually.
- New cases: In 2022, 924 new cases were recorded in children, an increase compared to previous years.
4.1.2 Diagnosis and treatment
- Diagnosis: The hemoglobin electrophoresis test is what diagnoses the disease, but many people are unaware of it or don’t have access to it, especially in the public health system.
- Diagnostic cost: The test in the private health system can range from 15 to 35 USD. Treatment is not subsidized by the government, although it is essential to control the symptoms of the disease.
4.1.3 Challenges
- Knowledge: There is a lack of awareness among teachers and family members about the severity of the disease, which makes it difficult to monitor students.
- Access to healthcare: The lack of resources in the public healthcare system exacerbates the situation, limiting access to proper diagnosis and treatment.
The main limitation in care is the concentration of specialized services in the country’s capital, with difficulties for patients in other provinces to access treatment due to financial and infrastructure constraints. The government is paying special attention to training health professionals for primary care, neonatal screening and comprehensive patient care, as well as raising awareness about voluntary blood donation. Several provinces in Angola lack hematologists to treat sickle cell anemia patients. This crisis has generated concern in society. It is a genetic and hereditary disease that causes the deformation of red blood cells. This altered form of red blood cells leads to a series of complications, such as chronic anemia and obstruction of blood vessels, resulting in pain (painful crises), a greater susceptibility to infections and damage to various organs over time. Diagnosis is made through blood tests. Treatments help control symptoms and prevent complications. This includes:
- Preventative antibiotics: To help prevent infections in children.
- Hydroxyurea: Medications that can help reduce the frequency of pain attacks.
- Blood transfusions: May be used in some cases.
- Folic acid supplementation: A vitamin that helps in the production of red blood cells.
It is essential that the diagnosis is made early, as adequate treatment significantly improves the quality of life of patients.
4.2 DISCUSSION
As we know, sickle cell anemia is a serious hereditary disease that significantly affects Angola, with estimates that around 20% of the population is carrier, which corresponds to more than three million people. The disease contributes to high infant mortality rates, with around 1,000 new cases annually and more than 1,000 child deaths recorded, due to complications such as vaso-occlusive crises, infections and organ damage. Access to adequate treatment, such as medication, blood transfusions, and testing, is limited in the public health system in many regions. In this regard, the Angolan government must make a major effort to improve its public policies to reduce the number of affected individuals, annual cases, and mortality. The government should pay special attention to training healthcare professionals for primary care, neonatal screening, and comprehensive patient care.
Sickle cell anemia is a serious and common hereditary disease in Angola, affecting a significant percentage of the population and causing several new cases annually. In this context, the government is making continuous, but insufficient, efforts to improve care for people with the disease, including the implementation of national public policies and professional training. The lack of government subsidies for treatment and widespread ignorance about the disease, including diagnostic tests, pose significant challenges.
5 Conclusions
We can conclude that the disease causes chronic anemia and pain crises. This disease is a public health challenge in Angola, affecting a significant portion of the population. Estimates indicate that approximately 20% of Angolans may carry the sickle cell trait, and approximately 1,000 children are born with the disease annually. Despite the high prevalence rate, diagnosis and treatment of the disease are hampered by a lack of resources, especially outside the capital, Luanda. Cooperation projects with other countries, such as Brazil, aim to support policy development and the training of Angolan health professionals to improve care and neonatal screening for the disease. It is the most prevalent genetic hematologic disease in the world, associated with high rates of morbidity and mortality. Sickle Cell Disease is a genetic and hereditary disease that causes the deformation of red blood cells. This altered form of red blood cells leads to a series of complications, such as chronic anemia and obstruction of blood vessels, resulting in pain (painful crises), a greater susceptibility to infections and damage to various organs over time. Diagnosis is made through blood tests, and treatment aims to control symptoms and prevent complications.
References
- Angola (2017). Ministério da saúde de Angola. Anemia falciforme. Disponível em https://www.anemiafalciforme-angola.org. Acessado 18 de Agosto de 2025.
- Grose, S. D.; Odame, I.; Atrash, H. K.; Amendah, D. D.; Piel, F. B.; WIlliams, T. N. (2011). Sickle cell disease in Africa: a neglected cause of early childhood mortality. Am. J. Prev. Med., v.41, n.6, suppl.4, p.S398-S405.-8733.2018v19n2p245-251.. http://dx.doi.org/10.17921/2447.
- Mggann, P. T.; Ferris, M. G.;Ramamurthy, U.; Santos, B.; Oliveira, V.; Bernardino, L.; Ware, R. E.(2013). A prospective newborn screening and treatment program for sickle cell anemia in Luanda, Angola. American Journal of Hematology, v.88, n.12, p.983-989, 2013.
- Oliveira, E. C. L.; Araújo, O. M. R. A.(2013). Aspectos clínicos da doença falciforme. In: IVO, M. L. (Org.) Hematologia: um olhar sobre a doença falciforme. Campo Grande, MS: Ed. UFMS, 2013. p.119-132. https://doi.org/10.1590/S1516-84842008000200012.
- Mousinho-Ribeiro, R. C., Cardoso, G. L.; Sousa, I. E. L.; Martins, P. K. C.(2008). Importância da avaliação da hemoglobina fetal na clínica da anemia falciforme. Rev. Bras. Hematol. Hemoter., v.30, n.2, p.136-141, 2008. https://doi.org/10.12957/reuerj.2020.51594.
- Sundd, P.,Gladwin, M. T.; Novelli, E. M (2019). Pathophysiology of sickle cell disease. Annu. Rev. Pathol., v.14, p.263-292. https://doi.org/10.1590/1982-0194201900028.
- Gould, J. (2005). Evaluating introgenic risk of youth suicide screening programs National Institute of Health Journal of the Amercan Medical Association.
- Creel, D., Witkop, C., King, R. (1974). Asymmetric visually evoked potentials in human albinos: evidence for visual system anomalies. Investigative Ophthalmology & visual science.1974, 13(6), 430-40.
- Simons, A. (2024) Chromosomes, Genes, and Traits: An Introduction to Genetics.
- Halder, R., Bridgeman-Shah, M. (1995). Skin cancer in African Americans – Wiley Online Library. Department of Dermatology, Howard University College of Medicine.
- Kruijt,C., Montoliu, L.(2022). The retinal pigmentation pathway in human albinism. National Institute Health, published in Progress in Retinal and Eye Research,
- Brücher, C., Peter, H.P., Grenzebach, U. (2019). Distribution of macular ganglion cell layer thickness in foveal hypoplasia: A new diagnostic criterion for ocular albinism. https://doi.org/10.1371/journal.pone.0224410.
- Hoffmann, M. (2025). Visual Processing Lab – Dpt Ophthalmology – Otto-von-Guericke-University Magdeburg.
- Schiaffino, M., Tacchetti, C. (2005). The ocular albinism type 1 (OA1) protein and the evidence for an intracellular signal transduction system involved in melanosome biogenesis. Doi:10.1111/j.1600-0749.2005.00240.x
- Keren, G., Lewis, C.(1993). A handbook for data analysis in the behavioral sciences: Methodological issues. Lawrence Erlbaum Associates, Inc…, 1993.
- Marjan, H., May, C. , Malicdan, J. (2020). Hermansky–Pudlak syndrome: Mutation update.. https://doi.org/10.1002/humu.23968
- Chiang, G.(2009). Effects of Feeding Solid-State Fermented Rapeseed Meal on Performance, Nutrient Digestibility, Intestinal Ecology and Intestinal Morphology of Broiler Chickens. Asian-Australasian Journal of Animal Sciences, 263-271. https://doi.org/10.5713/ajas.2010.90145.
- Mancini, P., Camilleri-Bröet, S., Anderson, B.(1998).. Proliferative mitochondrial dysfunction and apoptosis. https://doi.org/10.1016/S1566-3124(01)05005-2
- CRA (2023). Constituição da Republica de Angola, Órgão Oficial, I Série, Nº 154. 16 de Agosto de 2023, Artigo 77º (Saúde e Protecção Social).
- Lim, H., Kohli, I., Ruvolo, E. (2022). Impact of visible light on skin health: The role of antioxidants and free radical quenchers in skin protection. J Am Acad Dermatol 86(3S):S27-S37. Doi: 10.1016/j.jaad.2021.12.024.
- Lvovs, D., Favorova, O., Favorov, A.(2012). A Polygenic Approach to the Study of Polygenic Diseases”. 4 (3): 59–71. Doi:10.32607/20758251-2012-4-3-59 71. ISSN 20758251. PMC 3491892. PMID 23150804.
- Bick, D., Sarah, L., Dimmock, D. (2021). An online compendium of treatable genetic disorders”. American Journal of Medical Genetics. Part C, Seminars in Medical Genetics, 187 (1): 48–54. doi:10.1002/ajmg.c.31874. ISSN 1552-4876. PMC 7986124. PMID 33350578.
- Robinson, T. (2005).Genetics for Dummies (em inglês). Hoboken, NJ: Wiley Publishing, 2005, p. 9. 364 páginas. ISBN 978-0-7645-9554-7
- Willett, E. (2006). Genetics Desmystified. New York: McGraw-Hill.2006, p. 2-4. 210 páginas. ISBN 0-07-145930-8.
- Costa, P.N. (2019). Anemia falciforme, diagnóstico precoce e aconselhamento genético na doença falciforme: Uma revisão de literatura. Faculdade de Educação e Tecnologia da Amazonia, PA, Brasil. https://orcid.org/0009-0008-4434-1575.
- MINSA. (2023). Ministério da saúde de Angola. Anemia Falciforme. Disponível em https://www.gov.br/saude/pt-br/assuntos/saude-de-a-a-z/d/doenca-falciforme. Acessado 12 de Junho de 2025.
- MINSA (2023). Ministério da saúde. População Angolana com traço falciforme. Disponível em https://minsa.gov.ao/ e em http://www.anemiafalciforme-angola.org/. Acessado em 6 de Julho de 2025.
- Rees, D.C; Williams, T.N; Gladwin, MT. (2010). Sickle-cell disease. Lancet (London, England). 376 (9757): 2018–31. PMID 21131035. doi:10.1016/s0140-6736(10)61029-x.
- Wierenga, K. J.,Hambleton, I. R.,Lewis, N. A. (2001). Survival estimates for patients with homozygous sickle-cell disease in Jamaica: A clinic-based population study. Lancet 357 (9257): 680–683. PMID 11247552. Edit
- Kumar, V., Abbas, A.K.,Fausto, N.,Aster, J. (2009). Robbins and Cotran Pathologic Basis of Disease, Professional Edition: Expert Consult – Online (Robbins Pathology) (Kindle Locations 33530-33531). Elsevier Health. Kindle Edition.
- Adigwe, OP, Onoja, SO, Onaybayba, G (2023). A Critical Review of Sickle Cell Disease Burden and Challenges in Sub-Saharan Africa. J Blood Med, 14:367–376. doi: 10.2147/JBM.S406196Azevedo, E. S.(2010). Comentários sobre a descoberta do mecanismo de herança da anemia falciforme. Gazeta Médica da Bahia V.80; nº3 (3-5).Salvador, agosto-outubro de 2010 PDF Acesso Jan. 2015.
- Elzouki, A.Y. (2012). Textbook of clinical pediatrics 2 ed. Berlin: Springer. p. 2950.ISBN 9783642022012.
- Wierenga, K. J.; Hambleton, I. R.; Lewis, N. A. (2001). “Survival estimates for patients with homozygous sickle-cell disease in Jamaica: A clinic-based population study”. Lancet 357 (9257): 680–683. PMID 11247552. Edit
- Awasthy, N., Aggarwal, K. C., Goyal, P.C., Prasad,M.S., Saluja, S., Sharma, M. (2008). Sickle cell disease: Experience of a tertiary care center in a nonendemic area”. Annals of Tropical Medicine and Public Health 1 (1): 1–4. doi:10.4103/1755-6783.43069.