
Update in Diagnosis and Management of Interstitial Lung Diseases
Main Article Content
Abstract
Interstitial lung diseases (ILD) are a complex and diverse group of disorders. ILD are more frequently diagnosed and prevalent now. In this article, diagnosis approach, including new bronchoscopy and genetic tools, and some recently added concepts are revisited, as progressive fibrosing interstitial lung diseases and interstitial lung abnormalities.
Recently information relative to idiopathic pulmonary fibrosis is shown, including genetics and pathophysiology. We look over the dynamic world of interstitial lung diseases related to connective tissue diseases, principal characteristics of this group and the principles that define which of the various available therapies should be chosen. Finally new concepts and guidelines published about the diagnosis and management of hypersensitivity pneumonitis are reported. New data and treatments have changed our traditional vision of these lung diseases and we will new options in the next years.
Keywords: Lung Diseases, Interstitial; Idiopathic Interstitial Pneumonias; Idiopathic Pulmonary Fibrosis; Alveolitis, Extrinsic Allergic; Connective Tissue Diseases; hypersensitivity pneumonitis

Introduction
Interstitial lung diseases (ILD) are a diverse and challenging group of heterogeneous diseases. Idiopathic pulmonary fibrosis (IPF) is the most known but interstitial lung disease related to connective tissue diseases (C TD - ILD) and hypersensitivity pneumonitis (HP) have been more recognized and reported in the past years. There have been considerable advances in the last years, especially with new therapies and trials available.
In this review, we will update dia gnosis and therapies for interstitial lung diseases. Also, we revisited the last advances for more common diseases.
Diagnosis of interstitial lung diseases.
Precise diagnosis of interstitial lung disease is a challenging and probably the most difficult step in the study of these diseases.
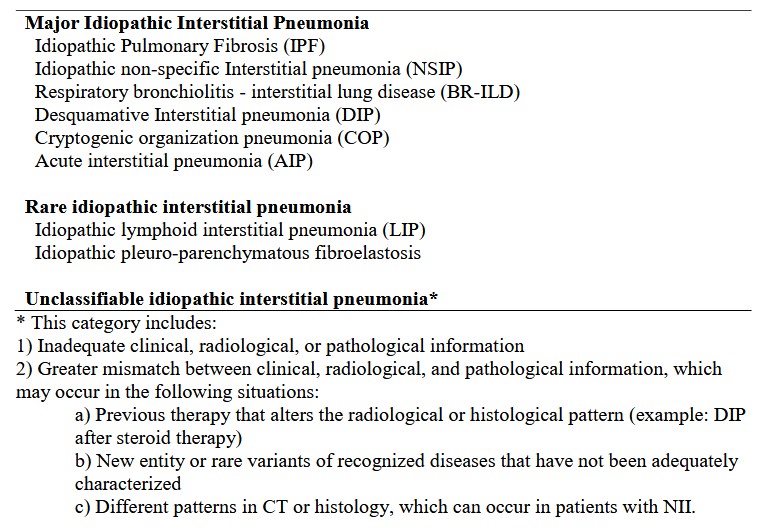
The last ATS / ERS consensus classification of idiopathic interstitial pneumonias (IIP) is presented in table 1 1 . High re solution computed tomography (HRCT) is the fundamental tool for diagnosis. Details about HRCT technique, lung images classification and histological findings have been published 2 . For usual interstitial pneumonia (UIP) pattern, usually the diagnosis and t herapeutic decisions will be straightforward.
In the scenery of diagnostic uncertainty after a exhaustive study, lung biopsy usually is proposed. Transbronchial lung cryobiopsy (TLC) as alternative to surgical biopsy is still controverted, with studies showing dissimilar results. The study of Romagnoli et al showed an important discordance between surgical biopsy and TLC 3 ; although, in the study of Troy et al TLC improved the ac curacy of diagnosis combined with other elements 4 . These apparently contradictory results could be merged in one sense; the sample size of TLC is bigger the sample size of surgical biopsy and the diagnosis hardly will be the same; moreover, we should not forget, the correlation between pathologist ILD diagnosis, using only surgical biopsies, is not good 5 . Therefore, is probably TLC add a small piece of information in many cases, but enough to allow the multidisciplinary committee take a therapeutic decision , as other studies have shown 6 . Complications of TLC as bleeding and pneumothorax are not uncommon and must be considered.
Table 1. Idiopathic interstitial pneumonia Classification 1
Recent evidence confirms than multidisciplinary committee improve the diagnosis accuracy of ILD 7 – 9 . Multidisciplinary committees (MDC) should summarize their work including diagnosis or working diagnosis, certainty about this diagno sis and proposed therapy 10 . Follow up is also, an important task for MDC. Initiatives to improve and standardize MDC are in progress and probably we will see new changes.
New genetic testing (Veracyte®, MUC5B promoter risk allele, Telomerase components ) to classify ILD are being reported 11 – 13 . Some of these, can be used in clinical setting today, but are not available in everywhere. The real - world efficacy of this tests is not clear yet, but some studies show promissory results 14 . As TLC, in a clinical context, can add certain at diagnosis. Studies in breath condensate are very promising 15 , especially for the simplicity to get the sample but they are still on development.
Emerging and evolving concepts.
Some new concepts have been added in the last years.
Interstitial pneumonia with autoimmune features (IPAF) concept was proposed in 2015 16 . IPAF is referred to patients with interstitial lung diseases and some findings related to autoimmune diseases, but do not fulfill criteria to a specific autoimmune condition. These findings have been divided in three categories: clinical domain (i.e.: rayna ud), serological domain (i.e.: a nti - cyclic citrullinated peptides) and morphological domain, which could be radiol ogical (i.e.: lymphoid interstitial pneumonia pattern) or histological (i.e.: organizing pneumonia combined with non - specific interstitial pneumonia). The concept of IPAF is not a specific diagnosis and there is still controversy about its usefulness 17,18
Progressive fibrosing interstitial lung diseases (PF - ILD) is another new concept. The use of a phenotype according to behavior of ILD was proposed many years ago 1 . The concept of PF - ILD has consolidated in the last years with trials showing effectiveness of therapy 19 . PF - ILD include non IPF diseases which show a sustained decline of lung function (forced vital capacity), imaging progression (fibrosis score) and/or clinical worsening (dyspnea). Specific criteria has been published 20 . Diseases i ncluded are idiopathic non - specific interstitial pneumonia, hypersensitivity pneumonitis, Sarcoidosis, unclassifiable interstitial pneumonias, CTD - ILD and others. The prevalence of PF - ILD could be a challenge for health system 21 . The INBUILD trial demonstr ated nintedanib use in these patients is effective 19 , with a magnitude of effect similar to that described inIPF patients. The use of pirfenidone shown positive effects in patients with unclassifiable interstitial pneumonia and progressive phenotype in one study, but there was controversial issues about methodology 22 . Other trials ongoing, including this kind of patients, will be available in next years.
Interstitial lung abnormalities (ILA) are referred to HRCT findings that are potentially compatible w ith ILD in asymptomatic patient. The concept of ILA is in evolution and there is not a universal definition. Patients with ILA should not have symptoms, physical exam findings or functional impairment; if these are present, it should be referred as mild di sease. ILA are increasingly recognized and some of these patients will evolve to IPF or other ILD, but there is not yet accuracy prediction tools 23 .
Idiopathic pulmonary fibrosis.
IPF is the most common ILD, at least in the north hemisphere countries, and the prevalence has increased. Is not clear, if this is by higher incidence or because the disease is more recognized 24 .
The IPF physiopathology is not completely understood yet, but has been advances in the last years. The MUC5B promoter risk allele is the most common mutation linked to IPF 25 . The MUC5B gene is related to mucociliary clearance epithelial activity but it is not clear how exactly the overexpression of mucin leads to develop IPF. Telomerase related mutations (TRM) have also been describ ed in patients with IPF, especially in familiar IPF. Different components of this protein complex can be affected for mutation related to impair the reparation function of telomerase. Some studies have found until 30 % of TRM in patients with familiar IPF and 10 % of TRM in patients with non - familiar IPF (sporadic) 26 .
Probably, the most notorious advance in the last year is the approval of drugs antifibrotic: nintedanib and pirfenidone. New evidence has shown both drugs can have an impact on survival 27,2 8 , something not proved in the initial trials. How long is extended this benefit and when to stop the treatment is not clear.
Trials of new promissory drugs and studies with combined drugs are ongoing (i.e., pirfenidone and nintedanib ), so is expected there will be new treatments in the next years. Non pharmacologic treatment components are very important, as rehabilitation, vaccines, and oxygen use. Gastroesophageal reflux disease treatment is controversial, with studies showing contr adictory results 29 – 32 . For patients with advanced diseases lung transplantation is the only option.
Connective tissue diseases associated with interstitial lung diseases.
The connective tissue diseases (CTD) that are often associated with interstitia l lung disease (ILD) include systemic sclerosis (SS), rheumatoid arthritis (RA), primary Sjogren's syndrome (pSS), idiopathic inflammatory myopathies (IIM), mixed connective tissue disease (MCTD) and systemic vasculitis. They may occur in patients with a k nown CTD or ILD may be the debut of the disease.
CTD - ILD are associated with significant morbidity and mortality 33 , however, compared to those with idiopathic interstitial pneumonias (NII), patients with CTD - ILD are more likely to respond to immunosuppressive therapy and have a better prognosis 34 .
All patients with CTD should be evaluated in targeted search for ILD and vice versa since the debut of symptoms and periodically thereafter. The evaluation should include a thorough clinical histo ry, physical exam, autoimmune serology, lung function tests and HRCT. Progressive dyspnea, cough and respiratory functional tests with restrictive pattern are common in CTD - ILD. Spirometry may be normal in mild illnesses. Gas diffusion capacity (DLCO) can be disproportionately reduced due to pulmonary hypertension (PH) or emphysema. For serial monitoring of patients with CTD - ILD, forced life capacity (FVC) and DLCO are frequently used to predict prognosis, progression and response to therapy 35 .
HRCT is more sensitive than chest x - ray and allows to describe the specific patterns that are associated with each CTD; the same patterns described for IIP are used (Table 1). The radiological pattern of NSIP is the most common in all CTD, except in RA where UIP p redominates 36 . In addition to the ILD pattern, HRCT provides information on the airways, pulmonary artery, pleura, pericardium, emphysema, presence of co - existing cancer and extra - pulmonary structures that may be relevant in the management of patient (i.e. : dilated esophagus, distal clavicular erosions).
A few years ago, a bronchioloalveolar lavage (BAL) rich in neutrophils or eosinophils was considered to represent an inflammatory pattern of CTD - ILD. There is currently consensus that BAL information do es not add value to lung function tests and HRCT findings to predict disease progression or response to therapy, except when lung infection is suspected 37 . Histopathology is not usually required in well - established cases of CTD - ILD 38 .
The distinction between IPF and CTD - ILD has important therapeutic implications. Antifibrotic agents such as pirfenidone and nintedanib have shown benefit in IPF and other progressive fibrous lung diseases, but immunomodulators such as azathioprine and prednisone, typically used in CTD - ILD , can be potentially harmful 39 . M ultidisciplinary discussion of these patients, including a trained rheumatologists, is essential to understand the differences in opportunity and aggressiveness of treatment, follow - up, prognosis, and timing for lung transplantatio n 40 .
Smoking cessation, pulmonary rehabilitation, oxygen sup plementation and appropriate vaccination, associated with the management of comorbidities such as gastroesophageal reflux disease (GERD), pulmonary hypertension (PH) and extrapulmonary manifestations of different CTD, make the integrated work of pneumology and rheumatology fundamental 41 .
Discrimination of the predominance of inflammatory versus fibrotic interstitial compromise in CTD - ILD commands therapeutic decisions. SS - ILD is the subgroup of CTD - ILD in which controlled randomized trials have been conducted. These treatments are used in other CTD for which there is not yet strong evidence. In 2016 was demonstrated the benefit in SS - ILD of mofetil mycophenolate (MMF), in a similar magnitude of effect to cyclophosphamide, with better tolerance and fewer adverse events 42 . Azathioprine, in routine clinical practice, is conside red a well - tolerated and commonly used alternative agent for maintenance therapy. In SS, corticosteroids should be avoided at doses higher than the equivalent of 15 mg of prednisone per day as it is associated with renal crisis 43 . Tocilizumab, an antibody against the interleucine - 6 receptor, was recently approved in SS - ILD patients with high skin sclerosis score and a systemic inflammatory profile 44 .
In the group of patients with fibrotic ILD, the utility of nintedanib, a triple tyrosine kinase inhibitor, h as been demonstrated to decrease the CVF rate of fall in patients with SS - ILD and in patients with other non - IPF progressive fibrosing diseases of autoimmune etiology (SS, RA) 19,45 . The effect of pirfenidone, the other antifibrotic agent approved in IPF, is currently being investigated in patients with SS - ILD with or without MMF 46
Autologous hematopoietic progenitor transplantation is another treatment that could be considered in patients with SS - ILD with rapidly progressive disease at risk of organ failure 47 .
Respect to RA disease modifying anti - rheumatic drugs, methotrexate may exceptionally produce acute pneumonitis, but its role in the development of pulmonary fibrosis has been definitively ruled out 48 . In RA - ILD there is active research with biological drugs, such as rituximab 49 , tocilizumab 50 , abatacept 51 and tofacitinib 52 .
Some patients can show a rapidly progressive ILD, particularly in patients with IIM. These potentially fatal ILD must be distinguished from chronic forms. Corticosteroids are the first therapeutic line in IIM - ILD. Rapidly progressive forms are usually treated with high - dose corticosteroids, associated with addition of a second or third immunosuppressant 53,54 . Calcineurin inhibitors (tacrolimus and cyclosporine A) have received special attention in patients with IIM - ILD 55 , although there are non - randomized controlled trials supporting these therapeutic decisions.
Hypersensitivity pneumonitis.
HP was not considered an important disease until early century 56 . In 2017, India ILD registry reported HP as the most common cause of ILD 57 . In USA, mortality has increased in the last 30 years 58 .
HP is an immune - mediated disease typically produced by inhalation of antigens. HP diagnosis is a challenge and there was no guidelines or consensus until last year. In 2020, it was published the first ATS Clinical Guideline of diagnosis of HP introducing changes and an algorithm to diagnosis 59 .
HP classically was divided in acute, subacute, and chronic without clear limits between them. The current classification propose two types: Fibrotic and No fibrotic. These types are relatively easy to define according to lung HRCT or lung biopsy. The guideline also defines the radiological signs and elements of HP, some these have not previously defined 59 .
The list of HP causes, and antigens is enormous, and every year are added new probable etiologies. Antigen identification is relevant and fail to identify the antigen is associated with worse prognosis 60 . Use of questionaries to identify potential antigen is suggested. Expert based qu estionaries has been published 61
The diagnosis is hard; HP must be considered in every new patient with ILD. Clinical course can be indolent or rapidly progressive and antigen is identified in about 50 % of patients. Some patients present as Flu - like symptoms. Patients can have digital clubbing and rales, more common in fibrotic form. HRCT can have a mix of ground glass opacities, air trapping areas and varied fibrotic changes 59 . Antigen and serological test are not standardized, and their interpretation can be misleading 62 . Lymphocytosis in BAL can be useful in some cases. Lung biopsy is a useful tool but may have interpretation challenges due to mix of patterns, especially when it is done by non - experienced pathologist 63 . Histopathology criteria has been published 59 .
The approach to diagnosis of HP will depend on the clinical presentation. In some cases, the history and Lung CT will be enough. Other patients will require BAL or lung biopsy. An expert panel report for diagnosis and evaluation has been p ublished 64 . Also an algorithm to classify the diagnosis confidence has been proposed 59 ( figure 1 ).
Therapy for HP is largely based in expert recommendations. Non fibrotic HP usually received treatment with corticosteroids and immunosuppressors, with variable results. There is not randomized controlled trials supporting these therapies and some observational studies suggest that these treatments could worsen the prognosis 65,66 . If an immunosuppressor treatment is started, you should follow up the patient closely to see improves or worsening and stop it.
For fibrotic HP, the study I NBUILD showed benefits on decline of FVC in patients with progressive phenotype 19 . In this trial, 25 % of sample had HP as diagnosis. If pirfenidone have the same effect, is not known.
Lung transplantation is an option in advanced disease.
Conclusions
In this review, we have summarized new information and concepts emerged in the field of ILD. It is not possible review every topic and we selected the more commons ILD with more new information available.
Although there is so much to learn about ILD, the knowledge about pathophysiology, genetics, clinical presentation, new laboratory tests, therapeutics ways and pharmacological alternatives have increased in the last decade. Multidisciplinary committees are consolidated, and they mu st be established as a standard on ILD management. Many trials on going about new drugs will be available in the next years and probably we will have new therapeutics alternatives.
Figure 1
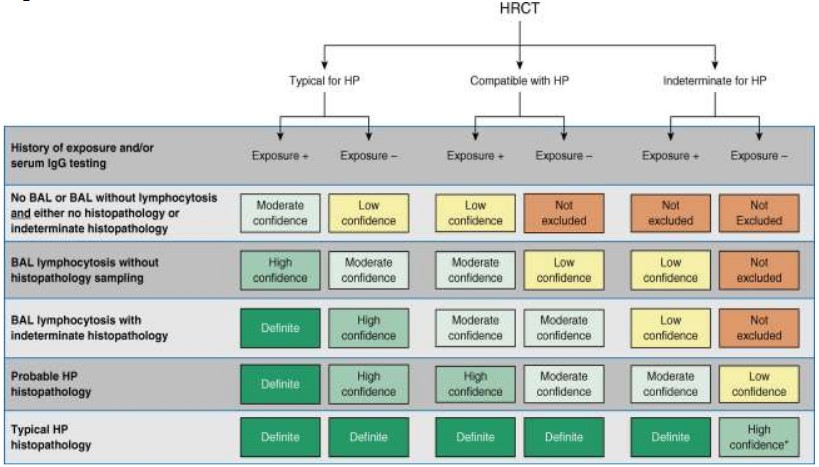
Figure 1. Hypersensitivity pneumonitis diagnosis based on incorporation of imaging, exposure assessment, BAL lymphocytosis, and histopathological findings. All confidence levels are subject to multidisciplinary discussion.
*Confidence may increase to “definite” if the pathologist’s conclusion persists after reevaluation in the context of additional clinical information or an expert second opinion on histopathology. HP = hypersensitivity pneumonitis; HRCT = high - resolution computed tomography.
Reprinted with permission of the American Thoracic Society. Copyright © 2021 American Thoracic Society. All rights reserved.
Raghu G, Remy - Jardin M, Ryerson CJ, Myers JL, Kreuter M, Vasakova M, Bargagli E, Chung JH, Collins BF, Bendstrup E, Chami HA, Chua AT, Corte TJ, Dalphin JC, Danoff SK, Diaz - Mendoza J, Duggal A, Egashira R, Ewing T, Gulati M, Inoue Y, Jenkins AR, Johannson KA, Johkoh T, Tamae - Kakazu M, K itaichi M, Knight SL, Koschel D, Lederer DJ, Mageto Y, Maier LA, Matiz C, Morell F, Nicholson AG, Patolia S, Pereira CA, Renzoni EA, Salisbury ML, Selman M, Walsh SLF, Wuyts WA, Wilson KC. 2020.
Diagnosis of Hypersensitivity Pneumonitis in Adults. An Offic ial ATS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 202(3):e36 - e69.
The American Journal of Respiratory and Critical Care Medicine is an official journal of the American Thoracic Society.
Article Details
The Medical Research Archives grants authors the right to publish and reproduce the unrevised contribution in whole or in part at any time and in any form for any scholarly non-commercial purpose with the condition that all publications of the contribution include a full citation to the journal as published by the Medical Research Archives.
References
2. Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of idiopathic pulmonary fibrosis An Official ATS/ERS/JRS/ALAT Clinical practice guideline. Am J Respir Crit Care Med. 2018;198(5):e44-e68. doi:10.1164/rccm.201807-1255ST.
3. Romagnoli M, Colby T V, Berthet J-P, et al. Poor Concordance between Sequential Transbronchial Lung Cryobiopsy and Surgical Lung Biopsy in the Diagnosis of Diffuse Interstitial Lung Diseases. Am J Respir Crit Care Med. 2019;199(10):1249-1256. doi:10.1164/rccm.201810-1947OC
4. Troy LK, Grainge C, Corte TJ, et al. Diagnostic accuracy of transbronchial lung cryobiopsy for interstitial lung disease diagnosis (COLDICE): a prospective, comparative study. Lancet Respir Med. 2020;8(2):171-181. doi:10.1016/S2213-2600(19)30342-X
5. Nicholson AG, Addis BJ, Bharucha H, et al. Inter-observer variation between pathologists in diffuse parenchymal lung disease. Thorax. 2004;59(6):500-505. doi:10.1136/thx.2003.011734
6. Tomassetti S, Wells AU, Costabel U, et al. Bronchoscopic lung cryobiopsy increases diagnostic confidence in the multidisciplinary diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2016;193(7):745-752. doi:10.1164/rccm.201504-0711OC
7. Walsh SLF, Wells AU, Desai SR, et al. Multicentre evaluation of multidisciplinary team meeting agreement on diagnosis in diffuse parenchymal lung disease: a casecohort study. Lancet Respir Med. 2016;4(7):557-565. doi:10.1016/S2213-2600(16)30033-9
8. Ageely G, Souza C, De Boer K, Zahra S, Gomes M, Voduc N. The Impact of Multidisciplinary Discussion (MDD) in the Diagnosis and Management of Fibrotic Interstitial Lung Diseases. Can Respir J. 2020;2020. doi:10.1155/2020/9026171
9. De Sadeleer LJ, Meert C, Yserbyt J, et al. Diagnostic Ability of a Dynamic Multidisciplinary Discussion in Interstitial Lung Diseases:
10. A Retrospective Observational Study of 938 Cases. Chest. 2018;153(6):1416-1423. doi:10.1016/j.chest.2018.03.026 10. Prasad JD, Mahar A, Bleasel J, et al. The interstitial lung disease multidisciplinary meeting: A position statement from the Thoracic Society of Australia and New Zealand and the Lung Foundation Australia *. Published online 2017. doi:10.1111/resp.13163
11. Raghu G, Flaherty KR, Lederer DJ, et al. Use of a molecular classifier to identify usual interstitial pneumonia in conventional transbronchial lung biopsy samples: a prospective validation study. Lancet Respir Med. 2019;7(6):487-496. doi:10.1016/S2213-2600(19)30059-1
12. Hunninghake GM, Hatabu H, Okajima Y, et al. MUC5B Promoter Polymorphism and Interstitial Lung Abnormalities. N Engl J Med. 2013;368(23):2192-2200. doi:10.1056/NEJMoa1216076
13. Stuart BD, Choi J, Zaidi S, et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat Genet. 2015;47(5):512-517. doi:10.1038/ng.3278
14. Kheir F, Alkhatib A, Berry GJ, et al. Using Bronchoscopic Lung Cryobiopsy and a Genomic Classifier in the Multidisciplinary Diagnosis of Diffuse Interstitial Lung Diseases. In: Chest. Vol 158. Elsevier Inc.; 2020:2015-2025. doi:10.1016/j.chest.2020.05.532
15. Moor CC, Oppenheimer JC, Nakshbandi G, et al. Exhaled breath analysis by use of eNose technology: A novel diagnostic tool for interstitial lung disease. Eur Respir J. 2021;57(1). doi:10.1183/13993003.02042-2020
16. Fischer A, Antoniou KM, Brown KK, et al. An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J. 2015;46(4):976-987. doi:10.1183/13993003.00150-2015
17. Lee JS, Fischer A. POINT: Does Interstitial Pneumonia With Autoimmune Features Represent a Distinct Class of Patients With Idiopathic Interstitial Pneumonia? Yes Chest. 2019;155(2):258-260. doi:10.1016/j.chest.2018.08.1074
18. Oldham JM, Danoff SK. COUNTERPOINT: Does Interstitial Pneumonia With Autoimmune Features Represent a Distinct Class of Patients With Idiopathic Interstitial Pneumonia? No. Chest. 2019;155(2):260-263. doi:10.1016/j.chest.2018.08.1073
19. Flaherty KR, Wells AU, Cottin V, et al. Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. N Engl J Med. 2019;381(18):1718-1727. doi:10.1056/nejmoa1908681
20. George PM, Spagnolo P, Kreuter M, et al. Progressive fibrosing interstitial lung disease: clinical uncertainties, consensus recommendations, and research priorities. Lancet Respir Med. 2020;8(9):925-934. doi:10.1016/S2213-2600(20)30355-6
21. Olson A, Hartmann N, Patnaik P, et al. Estimation of the Prevalence of Progressive Fibrosing Interstitial Lung Diseases: Systematic Literature Review and Data from a Physician Survey. Adv Ther. 2021;38(2):854-867. doi:10.1007/s12325-020-01578-6 22. Maher TM, Corte TJ, Fischer A, et al.
22. Pirfenidone in patients with unclassifiable progressive fibrosing interstitial lung disease: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Respir Med. 2020;8(2):147-157. doi:10.1016/S2213-2600(19)30341-8
23. Hatabu H, Hunninghake GM, Richeldi L, et al. Interstitial lung abnormalities detected incidentally on CT: a Position Paper from the Fleischner Society. Lancet Respir Med. 2020;8(7):726-737. doi:10.1016/S2213-2600(20)30168-5
24. Olson AL, Gifford AH, Inase N, Pérez ERF, Suda T. The epidemiology of idiopathic pulmonary fibrosis and interstitial lung diseases at risk of a progressive-fibrosing phenotype. Eur Respir Rev. 2018;27(150):180077. doi:10.1183/16000617.0077-2018
25. Hunninghake GM, Hatabu H, Okajima Y, et al. MUC5B Promoter Polymorphism and Interstitial Lung Abnormalities. N Engl J Med. 2013;368(23):2192-2200. doi:10.1056/nejmoa1216076
26. Courtwright AM, El-Chemaly S. Telomeres in interstitial lung disease: The short and the long of it. Ann Am Thorac Soc. 2019;16(2):175-181. doi:10.1513/AnnalsATS.201808508CME.
27. Dempsey TM, Sangaralingham LR, Yao X, Sanghavi D, Shah ND, Limper AH. Clinical effectiveness of antifibrotic medications for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2019;200(2):168-174. doi:10.1164/rccm.201902-0456OC
28. Nathan SD, Albera C, Bradford WZ, et al. Effect of pirfenidone on mortality: pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir Med. 2017;5(1):33-41. doi:10.1016/S2213-2600(16)30326-5
29. Johannson KA, Strâmbu I, Ravaglia C, et al. Antacid therapy in idiopathic pulmonary fibrosis: more questions than answers? Lancet Respir Med. 2017;5(7):591-598. doi:10.1016/S2213-2600(17)30219-9
30. Lee JS, Collard HR, Anstrom KJ, et al. Anti-acid treatment and disease progression in idiopathic pulmonary fibrosis: an analysis of data from three randomized controlled trials. Lancet Respir Med. 2013;1(5):369-376. doi:10.1016/S2213-2600(13)70105-X
31. Costabel U, Behr J, Crestani B, et al. Anti-acid therapy in idiopathic pulmonary fibrosis: insights from the INPULSIS® trials. Respir Res. 2018;19(1):167. doi:10.1186/s12931-018-0866-0
32. Tran T, Assayag D, Ernst P, Suissa S. Effectiveness of Proton Pump Inhibitors in Idiopathic Pulmonary Fibrosis: A Population-Based Cohort Study. Chest. 2021;159(2):673-682. doi:10.1016/j.chest.2020.08.2080
33. Marigliano B, Soriano A, Margiotta D, Vadacca M, Afeltra A. Lung involvement in connective tissue diseases: a comprehensive review and a focus on rheumatoid arthritis. Autoimmun Rev. 2013;12(11):10761084. doi:10.1016/j.autrev.2013.05.001
34. Jeganathan N, Sathananthan M. Connective Tissue Disease-Related Interstitial Lung Disease: Prevalence, Patterns, Predictors, Prognosis, and Treatment. Lung. 2020;198(5):735759. doi:10.1007/s00408-020-00383-w
35. Doyle TJ, Dellaripa PF. Lung Manifestations in the Rheumatic Diseases. Chest. 2017;152(6):1283-1295. doi:10.1016/j.chest.2017.05.015
36. Henry TS, Little BP, Veeraraghavan S, Bhalla S, Elicker BM. The Spectrum of Interstitial Lung Disease in Connective Tissue Disease. J Thorac Imaging. 2016;31(2):65-77. doi:10.1097/RTI.0000000000000191
37. Goh NSL, Veeraraghavan S, Desai SR, et al. Bronchoalveolar lavage cellular profiles in patients with systemic sclerosis-associated interstitial lung disease are not predictive of disease progression. Arthritis Rheum. 2007;56(6):2005-2012. doi:10.1002/art.22696
38. Lentz RJ, Taylor TM, Kropski JA, et al. Utility of Flexible Bronchoscopic Cryobiopsy for Diagnosis of Diffuse Parenchymal Lung Diseases. J Bronchology Interv Pulmonol. 2018;25(2):88-96. doi:10.1097/LBR.0000000000000401
39. Raghu G, Anstrom KJ, King TE, Lasky JA, Martinez FJ. Prednisone, Azathioprine, and N -Acetylcysteine for Pulmonary Fibrosis. N Engl J Med. 2012;366(21):1968-1977. doi:10.1056/NEJMoa1113354
40. Levi Y, Israeli-Shani L, Kuchuk M, Epstein Shochet G, Koslow M, Shitrit D. Rheumatological Assessment Is Important for Interstitial Lung Disease Diagnosis. J Rheumatol. 2018;45(11):1509-1514. doi:10.3899/jrheum.171314
41. Aparicio IJ, Lee JS. Connective Tissue Disease-Associated Interstitial Lung Diseases: Unresolved Issues. Semin Respir Crit Care Med. 2016;37(3):468-476. doi:10.1055/s-0036-1580689
42. Tashkin DP, Roth MD, Clements PJ, et al. Mycophenolate mofetil versus oral cyclophosphamide in sclerodermarelated interstitial lung disease (SLS II): a randomised controlled, double-blind, parallel group trial. Lancet Respir Med. 2016;4(9):708-719. doi:10.1016/S2213-2600(16)30152-7
43. Steen VD, Medsger TAJ. Case-control study of corticosteroids and other drugs that either precipitate or protect from the development of scleroderma renal crisis. Arthritis Rheum. 1998;41(9):1613-1619. doi:10.1002/1529-0131(199809)41:9<1613:AIDART11>3.0.CO;2-O
44. Khanna D, Lin CJF, Furst DE, et al. Tocilizumab in systemic sclerosis: a randomised, double-blind, placebocontrolled, phase 3 trial. Lancet Respir Med. 2020;8(10):963-974. doi:10.1016/S2213-2600(20)30318-0
45. Distler O, Highland KB, Gahlemann M, et al. Nintedanib for Systemic Sclerosis–Associated Interstitial Lung Disease. N Engl J Med. 2019;380(26):2518-2528. doi:10.1056/nejmoa1903076
46. Scleroderma Lung Study III - Combining Pirfenidone With Mycophenolate - Full Text View - ClinicalTrials.gov. Accessed March 28, 2021. https://clinicaltrials.gov/ct2/show/NCT 03221257
47. Sullivan KM, Goldmuntz EA, KeyesElstein L, et al. Myeloablative Autologous Stem-Cell Transplantation for Severe Scleroderma. N Engl J Med. 2018;378(1):35-47. doi:10.1056/nejmoa1703327
48. Juge P-A, Lee JS, Lau J, et al. Methotrexate and rheumatoid arthritis associated interstitial lung disease. Eur Respir J. 2021;57(2). doi:10.1183/13993003.00337-2020
49. Vadillo C, Nieto MA, Romero-Bueno F, et al. Efficacy of rituximab in slowing down progression of rheumatoid arthritis-related interstitial lung disease: data from the NEREA Registry. Rheumatology (Oxford). 2020;59(8):2099-2108. doi:10.1093/rheumatology/kez673
50. Manfredi A, Cassone G, Furini F, et al. Tocilizumab therapy in rheumatoid arthritis with interstitial lung disease: a multicentre retrospective study. Intern Med J. 2020;50(9):1085-1090. doi:10.1111/imj.14670
51. Fernández-Díaz C, Loricera J, Castañeda S, et al. Abatacept in patients with rheumatoid arthritis and interstitial lung disease: A national multicenter study of 63 patients. Semin Arthritis Rheum 2018;48(1):22-27. doi:10.1016/j.semarthrit.2017.12.012
52. Zhang J, Wang D, Wang L, et al. Profibrotic effect of IL-17A and elevated IL-17RA in idiopathic pulmonary fibrosis and rheumatoid arthritis-associated lung disease support a direct role for IL-17A/IL17RA in human fibrotic interstitial lung disease. Am J Physiol Lung Cell Mol Physiol. 2019;316(3):L487-L497. doi:10.1152/ajplung.00301.2018
53. Barba T, Fort R, Cottin V, et al. Treatment of idiopathic inflammatory myositis associated interstitial lung disease: A systematic review and metaanalysis. Autoimmun Rev. 2019;18(2):113-122. doi:10.1016/j.autrev.2018.07.013 Jablonski R, Bhorade S, Strek ME,
54. Dematte J. Recognition and Management of Myositis-Associated Rapidly Progressive Interstitial Lung Disease. Chest. 2020;158(1):252-263. doi:10.1016/j.chest.2020.01.033
55. Kurita T, Yasuda S, Oba K, et al. The efficacy of tacrolimus in patients with interstitial lung diseases complicated with polymyositis or dermatomyositis. Rheumatology (Oxford). 2015;54(1):39-44. doi:10.1093/rheumatology/keu166
56. Churg A, Ryerson CJ. The Many Faces of Hypersensitivity Pneumonitis. Chest. 2017;152(3):458-460. doi:10.1016/j.chest.2017.03.024
57. Singh S, Collins BF, Sharma BB, et al. Interstitial Lung Disease in India. Results of a Prospective Registry. Am J Respir Crit Care Med. 2017;195(6):801-813. doi:10.1164/rccm.201607-1484OC
58. Fernández Pérez ER, Sprunger D, Ratanawatkul P, et al. Increasing Hypersensitivity Pneumonitis-Related Mortality in the United States from 1988 to 2016. Am J Respir Crit Care Med. 2019;199(10):1284-1287. doi:10.1164/rccm.201807-1258LE
59. Raghu G, Wilson KC, Bargagli E, et al. Diagnosis of hypersensitivity pneumonitis in adults: An official ATS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2020;202(3):E36-E69. doi:10.1164/rccm.202005-2032ST.
60. Fernández Pérez ER, Swigris JJ, Forssén A V., et al. Identifying an Inciting Antigen Is Associated With Improved Survival in Patients With Chronic Hypersensitivity Pneumonitis. Chest. 2013;144(5):1644-1651. doi:10.1378/chest.12-2685
61. Petnak T, Moua T. Exposure assessment in hypersensitivity pneumonitis: a comprehensive review and proposed screening questionnaire. ERJ Open Res. 2020;6(3):230-2020. doi:10.1183/23120541.00230-2020
62. Kouranos V, Jacob J, Nicholson A, Renzoni E. Fibrotic Hypersensitivity Pneumonitis: Key Issues in Diagnosis and Management. J Clin Med. 2017;6(6). doi:10.3390/jcm6060062 63.
63. Trahan S, Hanak V, Ryu JH, Myers JL. Role of Surgical Lung Biopsy in Separating Chronic Hypersensitivity Pneumonia From Usual Interstitial Pneumonia/Idiopathic Pulmonary Fibrosis*: Analysis of 31 Biopsies From 15 Patients. Chest. 2008;134(1):126-132. doi:10.1378/chest.08-0033
64. Fernández Pérez ER, Travis WD, Lynch DA, et al. Diagnosis and Evaluation of Hypersensitivity Pneumonitis: CHEST Guideline and Expert Panel Report. Chest. Published online April 2021. doi:10.1016/j.chest.2021.03.066
65. Adegunsoye A, Oldham JM, Fernández Pérez ER, et al. Outcomes of immunosuppressive therapy in chronic hypersensitivity pneumonitis. ERJ open Res. 2017;3(3). doi:10.1183/23120541.00016-2017
66. Morisset J, Johannson KA, Vittinghoff E, et al. Use of Mycophenolate Mofetil or Azathioprine for the Management of Chronic Hypersensitivity Pneumonitis. Chest. 2017;151(3):619-625. doi:10.1016/j.chest.2016.10.029